生信媛
Linux shell trick for bioinformatics
(1)将一个文件按行倒序,将第一行变为倒数第一行,第二行变为倒数第二行
优雅至极的方法:tac yourfile.txt
稍微笨一点的方法:awk 'BEGIN{x=0}{x=x+1}{print x,$0}' yourfile.txt | sort -nr -k 1 | sed 's/^.*//g' | less
awk:给每一行标号x,格式变成:x 行内容
sort:按照第一列(行号)数值反向排序
sed:将开头的行号全局替代为空
less:好看地打印出来,使用者可以下拉阅读
(2)对于Rfam数据库下载下来noncoding RNAs的fasta文件,注释信息就在fasta文件的header里面。如果我们想要提取所有的sequence的注释信息。(注释信息以>开头,结果为注释信息去掉>)
grep '^>' rfam.fasta | tr -d ">" | less
(3)用Trinity组装之后得到的fasta格式的转录组文件,我们要做注释的话,有些header可能过长,所以我们要将header给截断些,仅保留最关键的信息。
cut -d " " -f 1 Trinity.fasta | less
(4)统计fastq的所有碱基的数目。
awk 'BEGIN{sum=0;]{if (NR%4==2){sum + = length($0);}}END{print sum;}' your.fq
sum:初始化碱基总数为0
NR%4:因为fastq的第二行才记录碱基信息,所以余数为2
| fastq格式 | |
|---|---|
| 第一行是header | |
| 第二行是碱基的排列顺序 | |
| 第三行是无用行 | |
| 第四行是与第二行相对应的质量值 |
length($0):length()函数求长度
(5)SAM文件中根据某一行的数值进行筛选,并且保留header,以拟南芥这种模式植物为例,和拟南芥的记忆组比对得到的SAM文件有9行header,我们想保留header,再根据某行值进行过滤。
awk '$3<1000 || NR <= 9' your.sam
筛选出第三列数值小于1000和前九行的数据。||表示或
注意用awk筛选的格式
(6)将fastq格式文件转化为fasta格式。
方法一:理解不了代替命令和N命令 sed '/^@/!d;s//>/;N' your.fastq > your.fasta
方法二:awk '{if(NR%4==1|| NR %4 ==2){print $0}}' your.fastq | sed 's/^@/>/g' | less
awk把fastq格式的第一行(header)和第二行(sequence)保留下来,再用替换命令将开头的@替换为>
(7) 当我们得到基因的注释之后,得到了two column的基因注释table, 但是里面有很多NA值。但我们不想看到NA,怎么办呢。
方法一:grep -v 'NA' file.txt
方法二:awk '{if(!/NA/){print $0}}' file.txt
方法三:sed -e '/*NA*/d' file.txt
方法四:awk '$0 !~ /NA/{print}' file.txt
(8)递归地查找当前目录下所有以".fasta"为后缀的文件,统计其行数。如果想统计fasta文件的所有碱基数目,请参考统计fastq碱基数目那一条。
find . -name "*.fasta" | xargs wc -l
xargs在里面的意义就是将fasta文件的名字,一个一个地传递给wc -l命令。
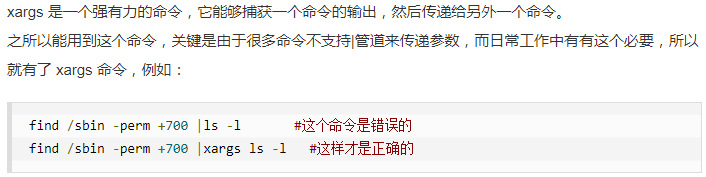
xargs一般是和管道一起使用的。命令格式是 somecommand | xargs -item command
(9)当你有两个gene list的文件,要找出仅在gene.file2中存在的行,不在gene.file1中出现的gene用来做GO分析。
grep -Fxv -f gene.file1 gene.file2
这里用grep比较两文件之间的不同,查过资料网上基本用的是 grep -Fvf
f指定的是范本文件file1,获取在file2但不在file1的记录
| options | description |
|---|---|
| -F | 将样式视为固定字符串的列表 |
| -x | 只显示全列符合的列 |
| -v | 反选 |
| -f file | 将file作为范本文件 |
(10)在某个目录下有很多文件,你想看看你最感兴趣的基因名字出现在哪个文件里,文件很多,并且子文件夹还有很多文件,即在一个目录下递归地查找匹配某字符串的文件。怎么办呢。
grep -Hrn "shengxinyuan"
| options | description |
|---|---|
| -H | with filename;在符合样式的行前,标示该行所属的文件名称 |
| -n | line number;在符合样式的行前,标示出该行的行列数 |
| -c | count;统计符合样式的列数(不要和-n搞混) |
| -r | recursive |
(??)有时候我们做一个clustalw,将多行的fasta(multiple line fasta)歌手文件,转行成单行的fasta文件(single line fasta)序列文件
awk '/^>/{print $0;};!/^>/{printf "%s",$0,n="n"}END{print ""}' test.fa
(12)go2geneID.txt文件中,GO号和gene之间是tab分开,gene之间是以逗号分开的。(awk中的split)
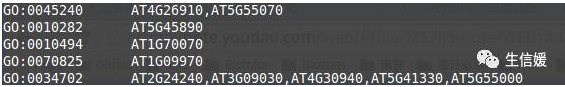
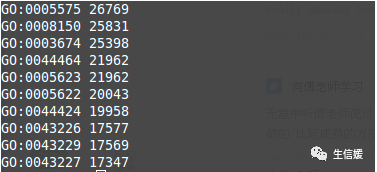
awk '{{split($2,array,",");split($0,table,"t");}{print table[0],length(arrary)}}' go2.geneID.txt | sort -nr -k 2 | head
结果如下图:基因数目最多的GO名称为GO:0005575,它有26769个基因
split作用:把一个字符串string按照field separator分割为单词并存储在数组array中。
格式 split (string,array,field separator)
split($2,array,",") 将gene按照逗号分隔开存储在array中([‘AT4G26910’,‘AT5G55070’])
split($0,table,"t") 将一行按照tab分割开存储在table中([‘GO:0045240’,‘AT4G26910,AT5G55070’])
table[0]:GO名称
length(array):计算一个GO名称的总基因数
sort -nr -k 2 :根据第二列(基因数)从大到小排序
(13)如何统计文件的行数
wc -l file.txt
awk 'END{print NR}' file.txt
(14)当我们想提取文件的某列时
cut -f 7 file.txt > out.txt
awk -F 't' '{print $7}' file.txt > out.txt
(15)将GFF里面第三列不是chromosome的行给取出来
awk '{if($3 != "chromosome){print $0}}' TAIR10.gff
(16)删除文件里所有的空行
sed '/^$/d' file.txt
(17)有时候我们不想看到header,直接用文件本身的内容去进行下一步操作,我们需要将header去掉。
sed '1d' file.txt
(18)有时候我们可能会需要使用awk为每一行行头添加字符串。
awk '{print "gene_ID" $0}' file.txt
(19)统计fastq文件里面的reads数目。
当质量行不以@开头时:grep -c "^@" test.fastq
当质量行以@开头时:cat test.fastq | echo $[ $(wc -l) /4 ]
(20)如何取出字符串的前50个字符
优雅的方法:cat your.fasta | grep -v ">" | cut -c 1-50
笨方法:cat your.fasta | grep -v ">" | sed 's/(.{50})*/1/g'
| cut -c 仅显示行中指定范围的字符 |
|---|
| n |
| n- |
| n-m |
| -m |
sed中运用了分组和引用分组的方法
(21)把一个Paired end序列的5’端前50个base和3’端的后50个base取出来做mapping
取出5’端前50个base:awk '{if(NR%4==2||NR%4==0){print substr($0,1,50)}else{print $0}}' your.fq
取出3’端的后50个base:awk '{if(NR%4==2||NR%4==0){print substr($0,length($0)-50+1)}else{print $0}}' your.fq
substr函数用于截取字符串
substr(s,p) 从s的第p个字符开始一直取到结尾
substr(s,p,n) 从s的第p个字符开始,取n个字符结束
if逻辑:对于一个fastq中的2,4行(sequence和quality)截取50个字符,1,3行(header和tag)原样保留
(22)去接头。用clustwl之类的工具,就能轻易找到sRNAs的adapter序列。
clustal sRNAs.fa
(23)去接头后,要去掉含有测序的ambiguous碱基,通常用N表示。如果序列中含有N碱基,这条reads就应该被抛弃。
sed 'N;s/n/t/' sRNAs.fa | sed -e '/t.*N/d' | tr "t" "n"
fasta是以两行为单位,一行是header,一行为sequence;N将下一行加入到模板空间,替换命令把两行变成一行(header t sequence)
将含有N的删掉(d),t只匹配sequence部分。
(24)去掉“N"这种ambiguous碱基之后,我们有必要做长度筛选,因为植物里面的sRNAs一般是18nt-30nt,而去掉adator之后,可能有些reads特别短了,还有些很长,可能是来自于structural RNAs的降解产物,所以应该去除这些过长或者过短的,只保留18-30的reads进行分析。
sed 'N;s/n/t/' test.fa | awk 'BEGIN{OFS="t"}{if(length($2)>=18 && length($2)<=30){print $1,"n",$2}}' | tr -d "t"
将header和sequence变为一行
用if语句筛选长度,然后先输出header加上换行符后,再输出sequence
用tr将sequence前面的分割符去掉
(25)对于去掉接头,去掉“N”碱基,去掉过短或者过长的sRNAs序列之后,我们下一步就该看看sRNAs有那些特征了,比如一个比较重要的特征就是5’端的碱基频率,因为不同的5’端碱基,决定了sRNAs可能与那种AGO蛋白相结合,然后发挥生理作用。
cat test.fa | grep -v ">" | cut -c 1-1 | sort | uniq -c | awk '{print $2,$1}' | tr " " "t"
uniq参数:
| option | description |
|---|---|
| -c | count;显示该行重复出现的次数 |
| -u | uniq;仅显示出现一次的行 |
| -d | repeated;仅显示重复出现的行 |
(??)sRNAs的长度分布
cat sRNAs.fa | awk 'NR%2==0' | awk '{print length($0)}'|sort |uniq -c|sed 's/^[][ ]*//g'|awk '{print $2,$1}'
(27) 递归地删除空目录
find . -depth -type d -empty -exec rmdir -v {} ;
| option | description |
|---|---|
| -empty | 空的文件 |
| -type d | 指定目录类型 |
eval–一层一层剥开我的心
eval CommandLine
eval对后面跟随的CommandLine进行两次扫描:第一次扫描会进行普通的命令执行和变量替换;
第二次扫描,若第一次扫描运行得到的命令或变量还可以再一次运行或替换,则再一次运行命令或进行变量替换。
(1)eval与变量二次替换
#定义两个变量
foo=10
x="foo"
y='$'$x
echo $y #显示$foo
eval y='$'$x
echo $y #显示10
(2)eval与命令执行
a="date"
eval `echo $a`
生信数据预处理的Linux三大神器
grep:最快的文本搜索工具
(1)以拟南芥基因组和注释文件作为联系对象
下载基因组文件
wget -c -4 -q http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas &
下载注释文件
wget -c -4 -q http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff &
(2)在注释文件中超找某个基因如AT5G25475
grep "AT5G25475" TAIR10_GFF3_genes.gff | head -n 5
(3)希望搜索AT5G25475但是不包含CDS
grep "AT5G25475" TAIR10_GFF3_genes.gff | grep -v "CDS" | head -n 5
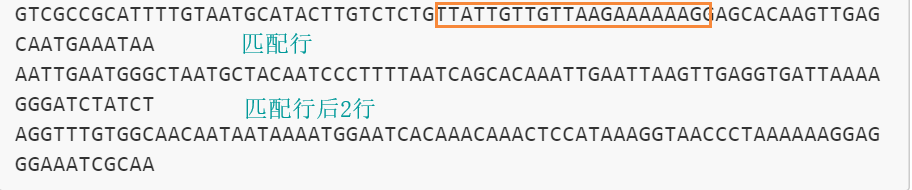
(4)基因组中查找某一段特定序列并查看上下文 -A(after) n:显示后n行
-B(before)n:显示前n行,-C= -A,-B
grep -A 2 'TTATTGTTGTTAAGAAAAAAGG' TAIR10_chr_all.fa
(5)在基因组统计某一序列出现的个数(-c:count)
grep -c 'TTATTGTTGTTAAGA' TAIR10_chr_all.fa
grep 'TTATTGTTGTTAAGA' TAIR10_chr_all.fa | wc -l
(6)只返回匹配到的内容(-o),注意使用-o参数不会返回完整的行
grep -o 'TTATTGTTGTTAAGA' TAIR10_chr_all.fa

(7)查找一个以AT5G254开头以1结尾的基因
grep 'AT5G254.*1$' TAIR10_GFF3_genes.gff
awk:强大的文本操作工具
awk逐行从文本中读取数据,将整行数据(record)定义为$0,然后根据指定的分隔符,将各列数据(record)分别定义为$1,$2,$3
格式:pattern1{action1};pattern2{action2};...
注意:如果没有定义pattern,则直接执行action;
如果没有提供action,则直接输出满足pattern的内容
(1)读出注释文件前两行
awk '{print $0}' TAIR10_GFF3_genes.gff | head -n 2
cat TAIR10_GFF3_genes.gff |head -n 2
(2)列出注释文件的1、4、5列
awk '{print $1,$4,$5}' TAIR10_GFF3_genes.gff | head -n 2
cut -f 1,4,5 TAIR10_GFF3_genes.gff
(3)awk还支持:算数运算(±*/%)逻辑运算(==,!=,>,<,>=,<=)或与非(!,||,&&)模式匹配(a~b, a!~b)
(4)找到长度大于10kb且在一号染色体的注释内容
awk '$5-$4>10000 && $1 ~/Chr1/' TAIR10_GFF3_genes.gff | head -n 5
(5)计算1号染色体长度cds的平均长度
awk 'BEGIN{sum=0;line=0};$5-$4>10000 && $1 ~ /Chr1/{sum+=($5-$4);line+=1};END{print "mean=" sum/line}' TAIR10_GFF3_genes.gff
(6)awk内部有许多特殊变量,如NR,表示当前所在的行数
显示第3-5行数据
cat TAIR10_GFF3_genes.gff | head -n 5 | tail -n 3
awk 'NR>=3 && NR<=5 {print $0}' TAIR10_GFF3_genes.gff
(7)
awk '$1~/Chr1/{feature[$3]+=1};END{for(k in feature) print k "t" feature[k]}' TAIR10_GFF3_genes.gff

(8)常用函数
| function | description |
|---|---|
| length(s) | s的长度 |
| tolower(s) | 转成小写 |
| toupper(s) | 转成大写 |
| substr(s,i,j) | 返回s的从i起始j个字符长度的部分 |
| split(s,a,d) | 根据分隔符d分割数据s,赋值给数组a |
| sub(f,r,s) | 根据正则f从r中提取数据到s |
sed:流处理工具
假如有一个文件a.txt如下文件:

(1)将chrom替换为chr
sed 's/chrom/chr/' a.txt
(2)只想打印第2行
sed -n '2p' a.txt
生信分析中基本Linux命令的使用
数据准备
下载拟南芥的参考基因组及其注释文件
wget http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas
wget http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff
file命令查看注释文件类型
file TAIR10_GFF3_genes.gff

了解数据内容
(1)查看文件前几行head
head -n 5 TAIR10_chr_all.fas
(2)查看文件后几行tail
tail -n 5 TAIR10_chr_all.fas
(3)逐页显示文本less
less TAIR10_chr_all.fas
一些小技巧
(1)显示文件前后几行
(head -n 2;tail -n 2) < TAIR10_chr_all.fas
(2)去除前面的comment line(有2行)
tail -n +2 file.gff
(3)调试管道命令(pipe)
对于管道命令的输出结果,可以及时使用less或者head查看,如果有错误可以及时用ctrl+c停止操作。
command1 | command2 | less
command1 | command2 | head -n
数据基本信息
(1)查看文本数据大小
-l:以长格式显示 -h:以G,M,K为单位显示数据大小
ls -lh TAIR10_chr_all.fa
(2)文件的行数
wc -l TAIR10_chr_all.fa
不统计注释行和无意义空白
grep -v "#" TAIR10_chr_all.fa | grep -v "^$" | wc -l
数据提取,排序和去重
(1)提取1,3,4,5列数据
cut -f 1,3,4,5 TAIR10_GFF3_genes.gff |head
保存为新文件
cut -f 1,3,4,5 TAIR10_GFF3_genes.gff | I() > part.txt
(2)根据第二列进行排序
sort -k2,2 part.txt | head -n 3
(3)先按照第一列逆序在根据第二列排序
sort -k1,1nr -k2,2 part.txt | head -n 3
(4)去重
cut -f 3 TAIR10_GFF3_genes.gff | sort | uniq -c

生信宝典
Linux学习-文件和目录
初识Linux系统
| section | description |
|---|---|
| ct | 用户名 |
| ehbio | 本机的名字;如果登录的是远程服务器,则为宿主机的名字 |
| ~ | 代表家目录 |
| $ | 用于指示普通用户输入命令的地方;对根用户来说一般用# |
ssh免密码登录远程服务器
原理:把本地电脑的公钥放在宿主机,然后使用本地电脑的私钥去认证。
最简单的操作
(1)安装/usr/bin/ssh-keygen -t rsa
安装完毕后查看~/.ssh目录会看到两个文件分别是 私钥 id_rsa 和公钥id_rsa.pub
(2)拷贝公钥到远程服务器的家目录下
scp ~/.ssh/id_rsa.pub user@remote_server:
(3)使用密码登录家服务器,执行
mkdir -p ~/.ssh; cat ~/id_rsa.pub>>~/.ssh/authorized_keys; chmod 700 ~/.ssh; chmod 600 >>~/.ssh/authorized_keys
Linux学习-文件操作
新建文件的方式:
| command | nano | vim |
|---|---|---|
| 新建文件 | nano filename | vim filename |
| 退出 | ctrl+x | Esc |
| 保存 | Y | :w |
| 进入写作模式 | 进入文件可直接写入 | i |
文件操作
#列出当前目录下有的文件和文件夹
ls
#新建一个文件夹
mkdir ehbio_project
#列出当前目录下有的文件夹及其子文件夹的内容
ls *
#拷贝data目录下的文件test.fa到ehbio_project目录下
cp data/test.fa ehbio_project/
#重命名data目录下的文件test.fa为first.fa
mv data/test.fa data/first.fa
# 进入另一个目录
cd ehbio_project
# 给文件做一份拷贝
cp test.fa second.fa
# 给文件多拷贝几次,无聊的操作,就是为了给rename提供发挥作用的机会
cp test.fa test2.fa
cp test.fa test3.fa
cp test.fa test4.fa
# 用rename进行文件批量重命名。根据test*.fa寻找匹配文件,在把这些文件名的中的test改为ehbio
rename 'test' 'ehbio' test*.fa
# 建立软连接,把当前目录下的ehbio2.fa,链接到上一层目录的data下面。建立软连接需要输入全路径
ln -s /home/ct/ehbio_project/ehbio2.fa ../data
Linux终端常用快捷操作
(1)命令或文件名自动补齐:输入命令或文件名的前几个字母后,按下Tab键,系统会自动补全或提示不全
(2)上下箭头:回溯之前的命令
(3)!string快速获取前面以string开头的命令
(4)ctrl+a回到命令的行首
写完命令突然不想执行:ctrl+a回到行首,在输入#,回车。命令就被注释掉了
(5)!!表示上一条命令
(6)对上一条命令进行替换 !!:gs// g为global,s为substitute
把上一个命令中全部ehbio替换为ehbio3
!!:gs/ehbio/ehbio3/
文件的可执行属性和环境变量
(1)which查看命令的路径
(2)文件属性rwx中,r表示read(4)、w表示write(2)、x表示execute(1)
三个为一组,连续出现三次。
给文件增加所有人可执行权限:chmod a+x file
给文件增加所有者可执行权限:chmod u+x file
给文件增加组内人或其它人可执行权限:chmod g+x,o+x file
表示拥有者有可读写执行权限,其它人有可读执行权限:chmod 755 file
exercise:
#新建一个文件
cat<<EOF>run.sh
echo "I am keiji"
EOF
#查看权限值
ls -l run.sh
#更改权限值
chmod 755 runsh
#去除其他用户的可执行权限
chmod o-x run.sh
#去除同组的可执行权限
chmod g-x run.sh
#给所有人增加可执行权限
chmod a+x run.sh
(3)系统环境变量名字为PATH,显示系统变量:echo $PATH
向环境变量中增加新路径时,路径必须是全路径。全路径指以/开头或已~开头的路径。
export PATH=$PATH:/home/ct
export命令只对当前终端有效,退出后就无效了。为了使得这一操作,长期有效,我们需要把这句话写入一个文件中~/.bash_profile

(5)因为系统查找命令是从PATH第一个目录到最后一个目录,第一次碰到查询的命令就会调用执行。如果系统存在一个python命令,我们现在又安装了一个python。如果想执行新安装的,就需要把/home/ct/anaconda/bin写在$PATH前面
export PATH=/home/ct/anaconda/bin:$PATH
Linux学习 - 文件内容操作(1)
(1)压缩文件:gzip 解压缩文件:gunzip
gzip -c将压缩的文件输出到STDOUT
gzip -c ehbio.fa > ehbio.fa.gz 相比gzip -v ehbio.fa 可以保留住原文件
解压缩文件gunzip ehbio.fa.gz
(2)wc -l获取文件行数
(3)获取文件中包含大于号的行 grep ">" ehbio.fa
统计包含大于号的行数 grep -c ">" ehbio.fa
删除end所在的的行 grep -v "END" ehbio.fa >ehbio2.fa
(4)去除HAHA
sed 's/HAHA//' ehbio.fa | head -n 4
(5)cut -f指定哪一列:取出第二列-f 2 取出第2-5列 -f 2-5 取出第2和第5列-f 2,5
cut -d指定分割符
Linux学习 - 管道、标准输入输出
(1)>表示标准输出 > filename将标准输出保存到文件filename中。标准错误还是显示在屏幕上。<表示标准输入
(2)2>&1 标准错误和标准输出同时输出
(3)- 短横线表示标准输出
(4)|表示把前一个命令的输出作为后一个命令的输入。
标准输出和标准错误都默认显示到屏幕上
bash stdout_error.sh
把结果输入到文件;标准错误显示在屏幕上
bash stdout_errot.sh > stdout_error.stdout
将错误和结果分别输入到文件中
bash stdout_errot.sh > stdout_error.stdout 2>stdout_error.stderr
标准输出和标准错误写入同一个文件
bash stdout_errot.sh > stdout_error.stdout 2>&1
将字符中的空格替换为换行。tr命令用于替换字符
echo "1 2 3" | tr ' ' 'n'
比较两个文件
#在第一个文件中存储内容
cat<<EOF>firstfile
content1
content2
EOF
#在第二个文件存储内容
echo "1 2 3" | tr ' ' 'n' > secondfile
#比较两个文件
diff firstfile secondfile
上述的操作可以通过一行命令来完成cat <<END | diff - <(echo "1 2 3" | tr ' ' 'n')
- (短横线)表示上一个命令的输出,传递给diff
< 表示其后的命令的输出,也重定向给diff
计算一行中的分段数
echo "actg aaaaa ccccg" | tr ' ' 'n' | wc -l
先输出行号再输出每行的内容
echo "a b c" | tr ' ' 'n' | sed '='
sed =:先输出行号,再输出每行的内容

echo "a b c" | tr ' ' 'n' | sed = | sed 'N;s/n/t/'
s: 替换

echo "a b c" | tr ' ' 'n' | sed = | sed 'N;s/^/>/'
N: 表示读入下一行;sed命令每次只读一行,加上N之后就是缓存了第2行,所有的操作都针对第一行;(偶数行相当于被隐藏了)
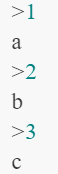
Linux学习-文件排序和FASTA文件操作
(1)环境变量的补充:
LD_LIBARY_PATH指定动态链接库(so文件)的位置
PYTHONPATH指定python安装包的路径
PERL5LIB指定perl的安装包路径
设置环境变量注意:设置时必须包含原始的环境变量,不能覆盖;注意设置目录顺序
(2)生成数字
seq:产生一系列数字 seq -s separator start (step) stop
产生1到10的数,步长为1 seq 1 10
产生1到10的数,步长为1,用空格分割 seq -s ' ' 1 10
生从1到10的数,步长为2 seq 1 2 10
cat<(seq 0 3 17)<seq(3 6 18)>test

(3)排序
sort默认按字符编码排序。
sort -n按数字大小排序
sort -u去除重复的行 相当于sort | uniq
sort file| uniq -c 获得每行重复的次数
要得到这个结果
sort test2 | uniq -c |awk 'BEGIN{OFS='t'}{print $2,$1}'

第二列按照数值排序 sort -k2,2n
第二列按照数值排序,再按第一列字母逆序排序 sort -k2,2n -k1,1r
(4)FASTA序列提取
生成单行序列的FASTA文件cat<<END>test.fa
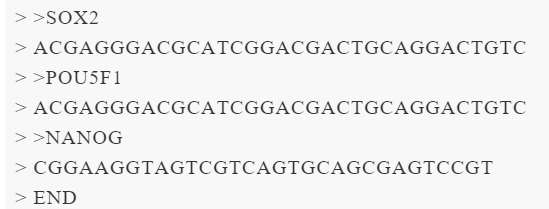 给>号开头的行的行尾加个TAB键,以便隔开名字和序列
给>号开头的行的行尾加个TAB键,以便隔开名字和序列
sed 's/^(>.*)/1t/' test.fasta

使用cat -A 可以显示文件中所有的符号 cat -A test.fasta
 把所有换行符替换为空格
把所有换行符替换为空格 sed 's/^(>.*)/1t/' test.fasta | tr 'n' ' '
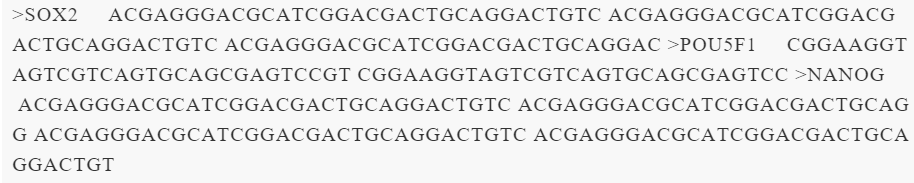
把最后一个空格替换为换行符
sed 's/^(>.*)/1t/' test.fasta | tr 'n' ' ' | sed -e 's/ $/n/' -e 's/ >/n>/g'
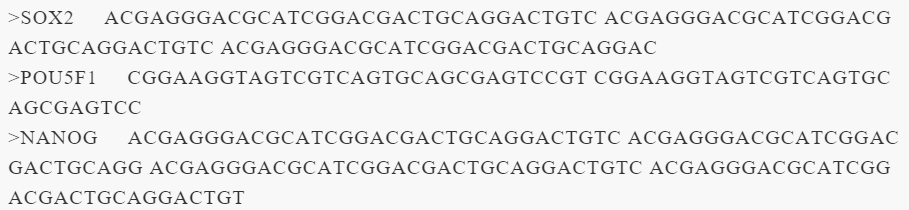
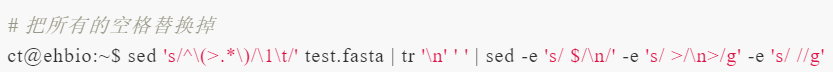

grep匹配含有SOX2的行,包含匹配的下一行
grep -A | grep 'SOX2' test.fasta
将多行FASTA序列转换为单行
cat file | sed 'N;s/n/t/' | tr 'n' ' '
sed 's/^(>.*)/1t/' test.fasta
Linux学习 - 命令运行监测和软件安装
(1)检测命令的运行时间 time command

(2)查看正在运行的命令或其资源使用 top
top -a按内存排序显示
top -u提示输入用户名,查看某个用户的进程
重点关注%MEM列,查看系统占用的内存是否超出。
查看系统进程ps auwx| grep 'process_name'
最后
以上就是美丽汽车最近收集整理的关于Linux生信笔记(公众号相关)1的全部内容,更多相关Linux生信笔记(公众号相关)1内容请搜索靠谱客的其他文章。







发表评论 取消回复