论文地址:Deep learning predicts tuberculosis drug resistance status from genome sequencing data
基于深度学习和基因组测序数据的结核病耐药性预测研究
作者信息:
[1]哈佛医学院生物医学信息系
[2]弗吉尼亚大学医学院
[3]分析机构
[4]关键路径研究所
[5]马萨诸塞州总医院肺科及危重科
概念介绍:
菌株:任何由一个独立分离的单细胞通过繁殖而成的纯遗传型群体及其后代。
MDR:对利福平[RIF]和异烟肼[INH]具有耐药性。
XDR: 对一种二线注射药物耐药,如阿米卡星[AMK]、卡那霉素[KAN]或卡波霉[CAP],以及一种氟喹诺酮耐药,如莫西沙星[MOXI]、氧氟沙星[OFLX]
一线用药:根据患者病情可以首先选择的药物。
二线用药:一线用药耐药以后选择的药物 。
表型:具有特定基因型的个体,在一定环境条件下所表现出来的性状特征的总和。
上位效应:一对基因显性基因的表现受到另一对非等位基因的作用。这种非等位基因间的抑制或遮掩作用叫上位效应。
研究背景:
结核病是全球十大死亡原因之一。抗生素的广泛使用导致耐药菌株的流行率增加。
据世界卫生组织估计,4.1%的新结核分枝杆菌临床分离株(MTB)是多药耐药(MDR)的,大约9.5%的MDR病例是广泛耐药的( XDR )。
48%的多药耐药结核病和72%的广泛耐药结核病患者有不良的治疗结果。
诊断药物的耐药性仍是提供适当结核病治疗的阻碍。
常规培养和基于培养的抗微生物药敏试验:结核分枝杆菌体外生长缓慢,构成了相当大的生物危害,需要数月才能报告结果。
分子诊断:世界卫生组织批准的三种分子测试,GenXpert、基于RT-PCR的快速检测(针对RIF)、LPA。但是它们仍存在缺陷。
1.灵敏度有限,依赖少数几个基因位点。
2.没有检测到大多数罕见的基因变异。
3.仅检测5种抗结核药物的耐药性。
4.没有考虑到遗传背景和基因-基因相互作用等变量。
全基因组测序:捕获了与耐药性有关的常见和罕见突变,成本低,速度快。但是,通过基因型数据预测表型的准确率仍与传统方法存在差距。
研究方法:
训练数据:汇集了来自世卫组织国家相关实验室和ReSeqTB知识库的数据。共包括 3,601个MTB分离株(其中1228株为多药耐药)。所有抗结核药物的敏感菌株比例均高于耐药菌株,不同药物的敏感菌株比例在53.0%至88.1%之间。
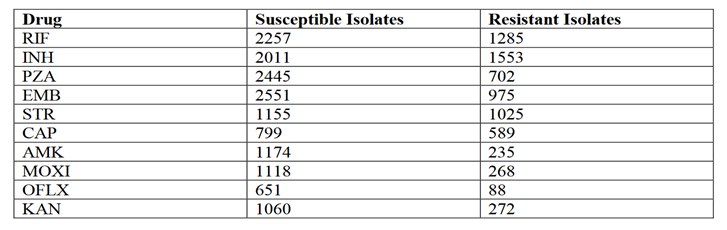
在MTB分离株基因组的30个启动子、基因间和编码区共发现了6342个不同的插入、缺失和单核苷酸多态性(SNPs)。 在这些变异中有166个,在3601个分离株中至少30个中出现。将其余的3445个变异聚合到141个派生类别中,并使用其中的56个变异作为预测因子,这些变异至少存在于30个分离株中。最终的模型在训练和后续分析中共使用了222个预测因子。
Tip:更新版论文将156->166。
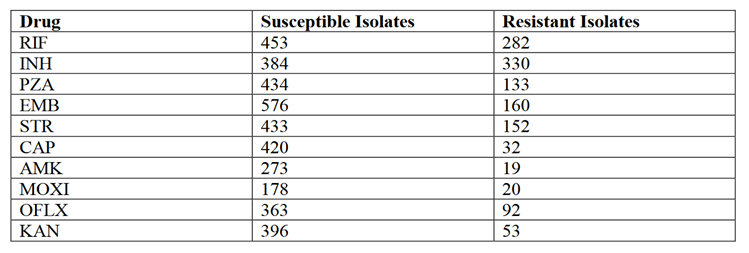
测试数据:收集了来自ReSeqTB的额外数据。 这些序列的表型抗性数据是可用的。包含了792个MTB分离株。由于环丙沙星在测试集中的表型有效性有限,且预测性能无法验证,所以没有考虑环丙沙星耐药性的性能。表型数据分类为耐药,敏感,或不可用。
MD-WDNN整体工作流程
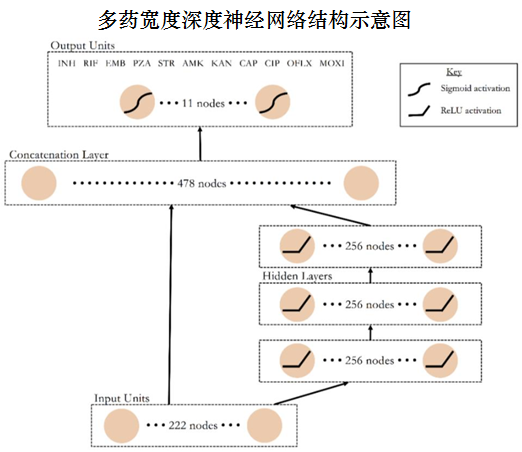
数据自下向上,通过神经网络的宽度(左)和深度(右)路径流动。在相应的节点上进行非线性转换。网络包含dropout、批标准化、L2正则化(应用在宽度部分)。优化器为Adam。
宽度深度神经网络(WDNN)结合了两种成功的模型,逻辑回归和深层多层感知器(MLP) ,允许网络从输入数据和更高层次的非线性特征中学习有用的规则。作者提出的多任务结构除了能够从只有部分表型的样本中学习之外,多药体系结构还共享了不同抗结核药物和基因的信息,以提供更准确的表型预测。
最后一层的11个节点代表每一种药物,其输出值为MTB分离株对相应药物产生耐药性的概率。
作者构建了单药WDNN (kSD-WDNN和SD-WDNN),除了输出层的结构是对一种药物进行预测外,其结构与多药模型相同。
损失函数: MD-WDNN利用的损失函数是传统二元交叉熵的一种变体。由于数据集缺少一些药物的MTB分离株的耐药性数据,所以作者实现了一个损失函数,该函数不惩罚模型对没有表型数据的药物-分离株对的预测。 由于一些药物的敏感性和耐药性类别不平衡,作者根据每种药物中的敏感-耐药比率调整了损失函数,增加了稀疏类别的权重。
因此,最终的损失函数是一个类权重的二元交叉熵,它掩盖了在没有耐药状态的情况下的输出。
为了预测每株分枝杆菌的耐药性,作者建立了一个多药(多任务)宽度深度神经网络(MD-WDNN)来同时预测所有药物的表型状态。
比较实验包括:
(1)一个单任务的WDNN,其中每种药物都是使用一个单独的模型进行训练(SD-WDNN)。
(2)仅使用常见突变训练的MD-WDNN。
(3)一个没有“宽度”部分的多层感知器(深层MLP )。
(4)根据预先选择的基因突变训练的单一药物WDNN(kSD-WDNN)。
(5)单一药物随机森林模型
(6)单一药物带L2正则的逻辑回归模型
所有的比较都使用训练数据进行10折的交叉验证,重复5次,总共50个模型。 最终的模型(仅MD-WDNN)性能通过一个独立的验证集进行验证。
t-SNE降维(t-分布随机邻域嵌入)
是一种高维数据的无监督非线性降维算法。
基本原理:
SNE是通过变换将数据点映射到概率分布上,主要包括两个步骤:
(1)SNE构建一个高维对象之间的概率分布,使得相似的对象有更高的概率被选择,而不相似的对象有较低的概率被选择。
(2)SNE在低维空间里在构建这些点的概率分布,使得这两个概率分布之间尽可能的相似。
作者使用t分布随机近邻嵌入(t-SNE)可视化并进行特征重要性分析以探究药物间的相似性。
实验结果:
MTB分离株多样性评估
使用33个遗传谱系标记的序列数据,计算分离株之间的欧式距离。 构造系统树图并将这些分离株分为五个集群,与MTB已知的遗传谱系相对应。
其中632个来自欧美拉丁美洲地中海亚系,1501个来自其他欧美亚系,331个来自印度洋或非洲,643个来自中亚,494个来自东亚。

分离株遗传谱系结构的影响
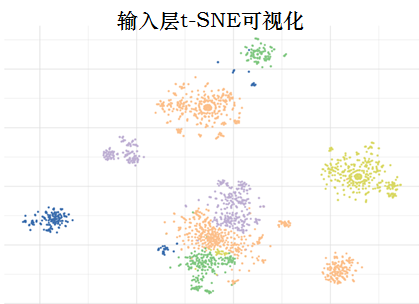

左图在很大程度上再现了血统导致的遗传聚类,说明分离株之间最大的遗传差异与血统有关。
右图证实了MD-WDNN对表型的预测不受谱系相关变异的影响。
意义: 通过比较输入数据和模型预测的t-SNE可视化,证实了该模型不受分离株遗传谱系结构的影响,而这正是MTB基因型和表型关系公认的混淆因子。
验证和测试结果
验证:
作者列出了所有模型10折交叉验证,重复5次,共50个模型得到的AUC结果。
根据AUC对10种抗结核药物进行了从1-7(最好到最差)的排名,并报告了每个模型的平均排名。
测试:
对于MD-WDNN,在额外的验证集上验证了模型的性能。进行了两组实验。
第一组,通过确定概率阈值,以最大化每种药物的特异性和敏感性之和。
第二组,确定概率阈值,使敏感性最大化且给定的特异性至少为90%。
90%的特异性阈值来源于价值评估,由于治疗毒性和副作用,过度诊断抗生素耐药性比诊断不足更有害。例如会造成肾衰竭和听力损失。
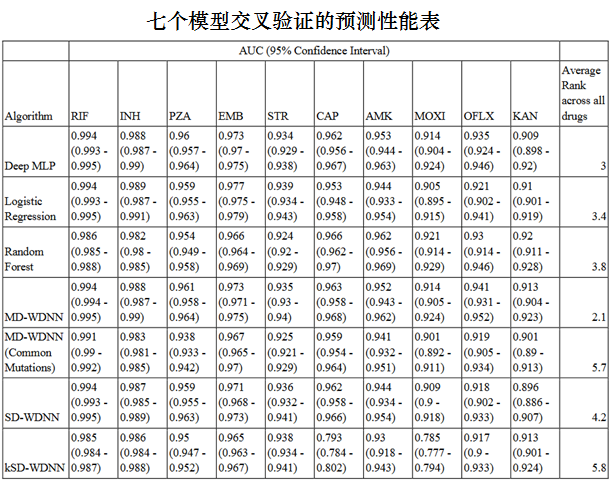
将MD-WDNN、SD-WDNN、deep MLP、random forest和logistic回归模型在全组预测因子上进行训练。MD-WDNN(常见变异)是针对不包括衍生类别在内的变异进行训练的,而kSD-WDNN是针对已知是每种药物耐药性决定因素的预选突变进行训练的。表显示了交叉验证得到的平均AUC和95%置信区间。
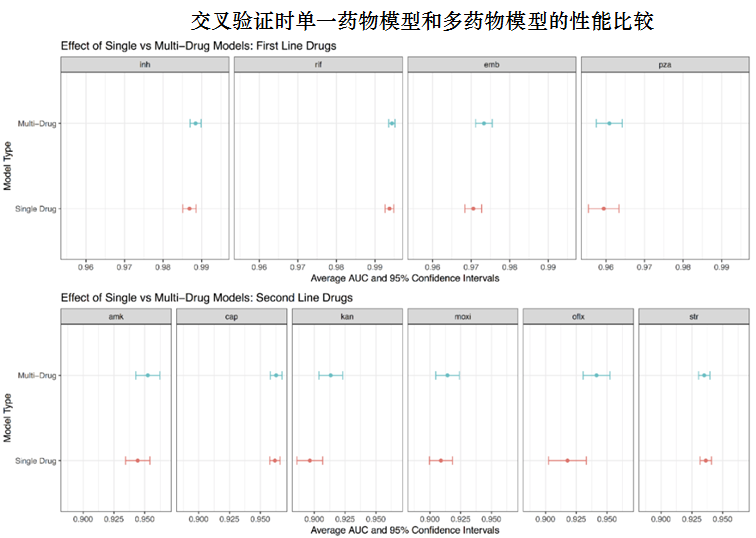
SD-WDNN一线药物的平均AUC为0.978,二线药物为0.928。
MD-WDNN一线药物的平均AUC为0.979,二线药物为0.936。
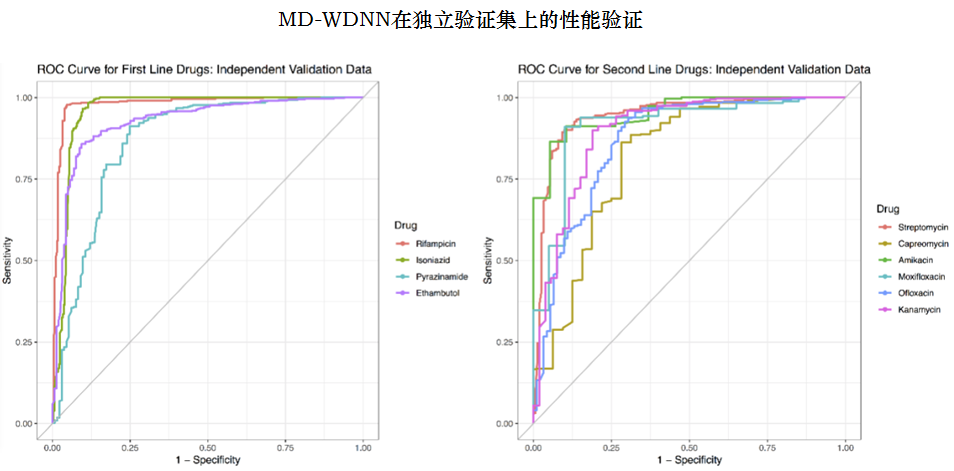
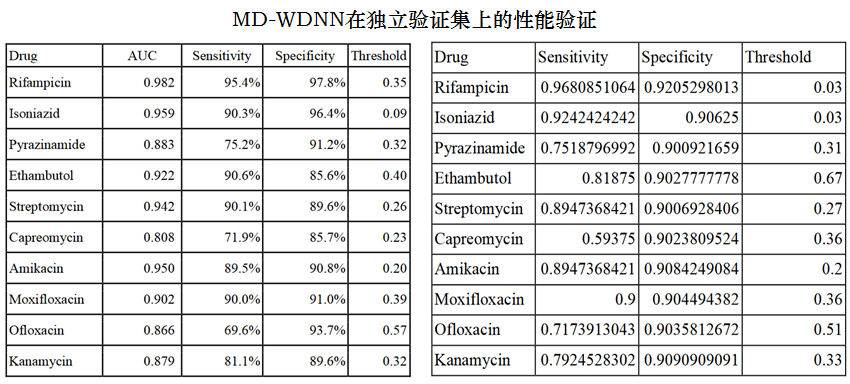
一线药物平均AUC为0.937,二线药物平均AUC为0.891。
一线药物的平均灵敏度和特异度为87.9%和92.7%,二线药物的平均灵敏性和特异度为82.0%和90.1%。以上数据针对左表。
在左表中选择的阈值最大化灵敏度和特异性之和。
右表中选择的阈值在特异度至少为90%的情况下使灵敏度最大化。
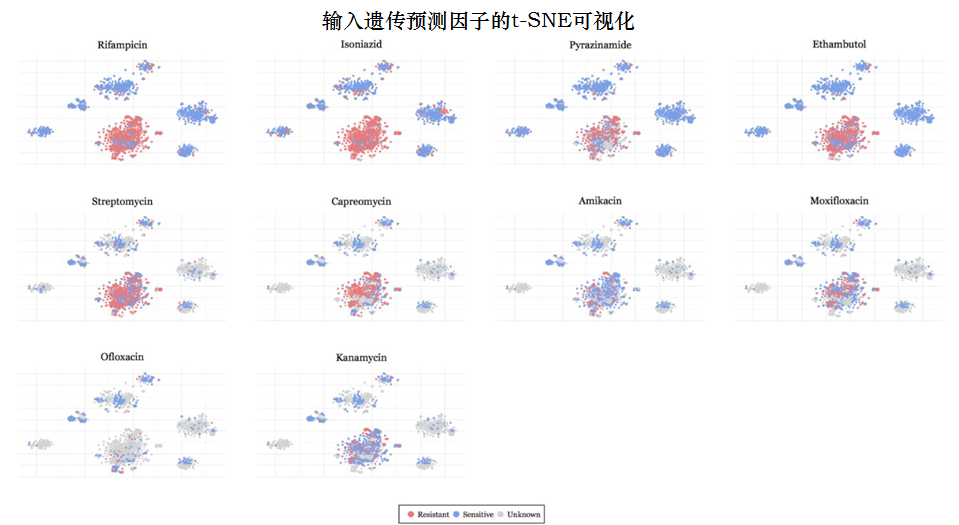
输入的遗传标记最初在222维空间中,投射到二维空间中。每个点是一个MTB分离株。输入遗传标记上的t-SNE表现出明确的簇状结构,簇间的抗性分类模式难以识别。
对10种药物所有222个预测因子通过置换实验来测试对耐药状态的重要性。一线抗结核药物具有最多数量的重要“耐药预测因子”:利福平(143个预测因子)、异烟肼(144个预测因子)、吡嗪酰胺(132个预测因子)、乙胺丁醇(140个预测因子) 。
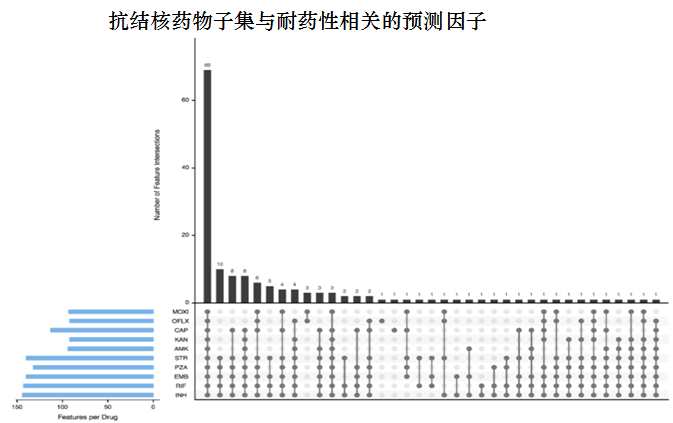
下图显示了每种药物的重要预测因子的数量和每个药物子集的突变数量。有37个药物子集共享至少一个耐药预测因子。最大的一个子集是10种抗结核药物,共有69种耐药性预测因子。
作者置换了抗性标签并计算了差异的分布,P(分离株耐药|有突变)-P(分离株耐药|无突变)。
MTB基因变异对耐药预测的重要性
作者对222个预测因子均进行了置换测试。对每个预测因子,进行了100000次随机置换。最终的差异评估显著性水平α= 0.05。
由实验得到的生物学结论:
包含一种第二行注射药物并且包含至少两个预测因子的药物子集一致包含INH和RIF。这与先前知识一致,即在一线药物有耐药性后,才会使用二线药物。多药模型能够捕捉到这些关系。
氟喹诺酮类药物子集共有3个耐药性相关预测因子,这些预测因子在其他一线或二线药物中均未发现,表明氟喹诺酮类药物具有不同于其他药物的作用机制。
实现细节
WDNN和MLP模型实现使用Python 2.7中的Keras 1.2.0库,后端是TensorFlow 0.10.0。用Python Scikit-Learn 0.18.1实现了随机森林和正则化逻辑回归分类器。采用R3.4.0实现分离株多样性分析,使用R中的Rtsne 0.13包实现t-SNE分析,置换测试用Python 2.7实现。所有模型都是在NVIDIA GeForce GTX Titan X图形处理单元(GPU)上进行训练的。

主要贡献:
本研究的主要目的是利用基因组数据建立一个高度精确的耐药性预测模型。
(1)作者将30个基因位点集合中的所有变异作为每种药物耐药性的潜在预测因子,并没有根据先验知识将变异分类。这种更加“宽容”的方法所带来了可观的性能提升,特别是对于二线药物和一线药物吡嗪酰胺。
(2)多药结构允许表型数据较少的药物从分离株表型数量较多的药物中借用信息。
(3)宽度和深度的结构有利于获取关于MDR和XDR遗传病因的先验信息。网络的宽度部分使变异的影响很容易学习,而网络的深层部分允许任意复杂的上位效应影响预测。
本文提出的模型是第一个可以同时预测10种抗结核药物的耐药性的多任务工具,同时具有最先进的性能。
不足之处:
(1)表型数据可能存在偏差,因为一些药物的药敏试验已被证明具有低重现性和高方差。
除了测试的技术和实验室限制外,某些耐药性突变,特别是乙胺丁醇和二线药物,可能导致最低抑制浓度(MICs)非常接近临床试验浓度,这可能导致预测的敏感性和特异性较低[1]。使用MIC数据构建未来的学习模型可能有助于克服这一点。
(2)模型只包括单独发生的概率>0.8%(3601个分离株中的30个)的突变,或与同一基因或基因间区域的其他罕见突变聚集在一起时发生的突变。可能漏掉了一些重要的预测因子。
(3)由于缺乏表型数据,本文没有包括环丝氨酸、对氨基水杨酸等三线抗结核药物。
[1]Ängeby K, Juréen P, Kahlmeter G, Hoffner SE, Schön T. Challenging a dogma:antimicrobial susceptibility testing breakpoints for Mycobacterium tuberculosis. Bull World Health Organ. 2012;90(9):693–8.
总结:
深度学习中的多任务架构在药物相关行业中没有得到广泛的应用,因为存在很多障碍,例如实现高质量的深度多任务网络比较困难。当使用更小的数据集时,多任务神经网络常比单个任务模型有更大的性能增益。
多药框架虽然对几种药物的效果不明显,但卡那霉素和氧氟沙星的效果提升显著。这是一项重要进展,因为二线注射剂和氟喹诺酮类药物是MDR-TB治疗的基础药物,迄今为止,在使用简单直接关联方法时,对二线药物耐药性的预测受到有限的遗传知识库和因此有限的敏感性的挑战。因此,使用更复杂的模型,如MD-WDNN,对于一部分抗结核药物来说是合理的。
深度学习模型可以随着数据集的更迭而快速更新。预计随着更多的数据的纳入,二线注射药物和氟喹诺酮类药物的敏感性和特异性差距将会减小。
最后
以上就是任性心锁最近收集整理的关于生物信息学之抗癌药物反应论文阅读四:MD-WDNN的全部内容,更多相关生物信息学之抗癌药物反应论文阅读四内容请搜索靠谱客的其他文章。








发表评论 取消回复