继续上篇文章所做的,接下来进行数据质控使用R语言代码:
scRNA <- scRNA3 #以后的分析使用整合的数据进行
##meta.data添加信息
proj_name <- data.frame(proj_name=rep("demo2",ncol(scRNA)))
rownames(proj_name) <- row.names(scRNA@meta.data)
scRNA <- AddMetaData(scRNA, proj_name)
##切换数据集
DefaultAssay(scRNA) <- "RNA"
##计算线粒体和红细胞基因比例
scRNA[["percent.mt"]] <- PercentageFeatureSet(scRNA, pattern = "^MT-")
#计算红细胞比例
HB.genes <- c("HBA1","HBA2")
HB_m <- match(HB.genes, rownames(scRNA@assays$RNA))
HB.genes <- rownames(scRNA@assays$RNA)[HB_m]
HB.genes <- HB.genes[!is.na(HB.genes)]
scRNA[["percent.HB"]]<-PercentageFeatureSet(scRNA, features=HB.genes)
#head(scRNA@meta.data)
col.num <- length(levels(as.factor(scRNA@meta.data$orig.ident)))
##绘制小提琴图
#所有样本一个小提琴图用group.by="proj_name",每个样本一个小提琴图用group.by="orig.ident"
violin <-VlnPlot(scRNA, group.by = "proj_name",
features = c("nFeature_RNA", "nCount_RNA", "percent.mt","percent.HB"),
cols =rainbow(col.num),
pt.size = 0.01, #不需要显示点,可以设置pt.size = 0
ncol = 4) +
theme(axis.title.x=element_blank(), axis.text.x=element_blank(), axis.ticks.x=element_blank())
ggsave("cluster1/vlnplot_before_qc.pdf", plot = violin, width = 12, height = 6)
ggsave("cluster1/vlnplot_before_qc.png", plot = violin, width = 12, height = 6)
plot1 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot3 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "percent.HB")
pearplot <- CombinePlots(plots = list(plot1, plot2, plot3), nrow=1, legend="none")
ggsave("cluster1/pearplot_before_qc.pdf", plot = pearplot, width = 12, height = 5)
ggsave("cluster1/pearplot_before_qc.png", plot = pearplot, width = 12, height = 5)
##设置质控标准
print(c("请输入允许基因数和核糖体比例,示例如下:", "minGene=500", "maxGene=4000", "pctMT=20"))
minGene=500
maxGene=3000
pctMT=10
##数据质控
scRNA <- subset(scRNA, subset = nFeature_RNA > minGene & nFeature_RNA < maxGene & percent.mt < pctMT)
col.num <- length(levels(as.factor(scRNA@meta.data$orig.ident)))
violin <-VlnPlot(scRNA, group.by = "proj_name",
features = c("nFeature_RNA", "nCount_RNA", "percent.mt","percent.HB"),
cols =rainbow(col.num),
pt.size = 0.1,
ncol = 4) +
theme(axis.title.x=element_blank(), axis.text.x=element_blank(), axis.ticks.x=element_blank())
ggsave("QC/vlnplot_after_qc.pdf", plot = violin, width = 12, height = 6)
ggsave("QC/vlnplot_after_qc.png", plot = violin, width = 12, height = 6)质控后的结果: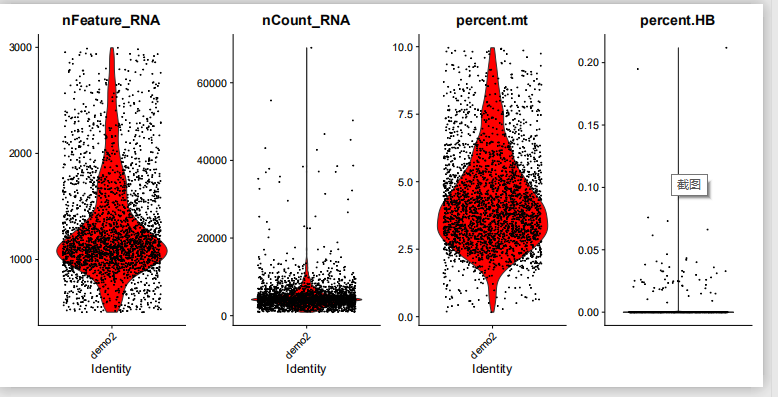 质控前的结果:
质控前的结果:
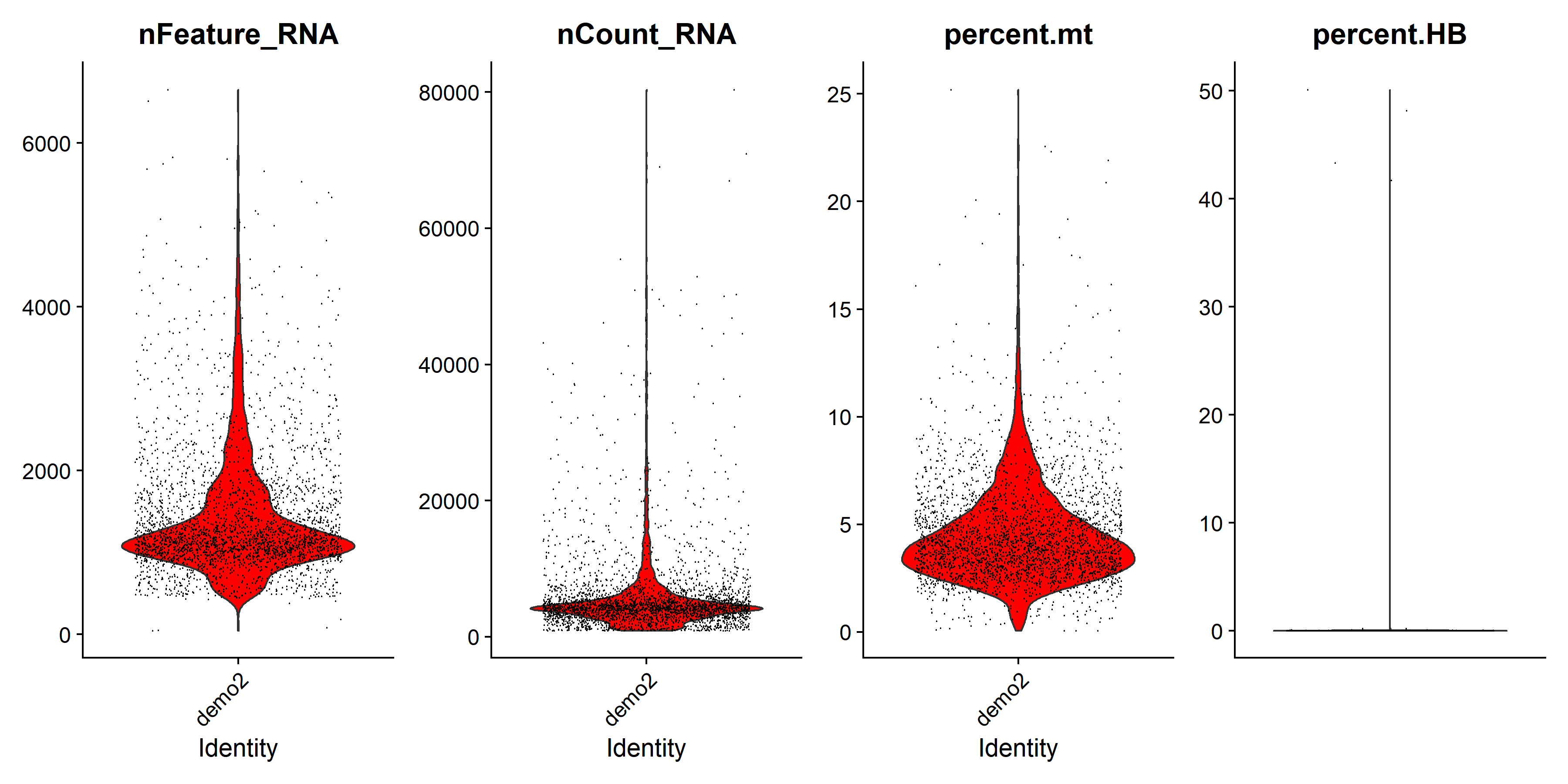
前后对比很明显,这也就说明了对数据的质控的重要性。
最后我所做的所有分析与教程的代码都会在我的个人公众号中,请打开微信搜索“生信学徒”进行关注,欢迎生信的研究人员和同学前来讨论分析。
ps:公众号刚刚建立比较简陋,但是该有的内容都不会少。
最后
以上就是有魅力菠萝最近收集整理的关于单细胞多样本多平台合并分析(二)的全部内容,更多相关单细胞多样本多平台合并分析(二)内容请搜索靠谱客的其他文章。
本图文内容来源于网友提供,作为学习参考使用,或来自网络收集整理,版权属于原作者所有。








发表评论 取消回复