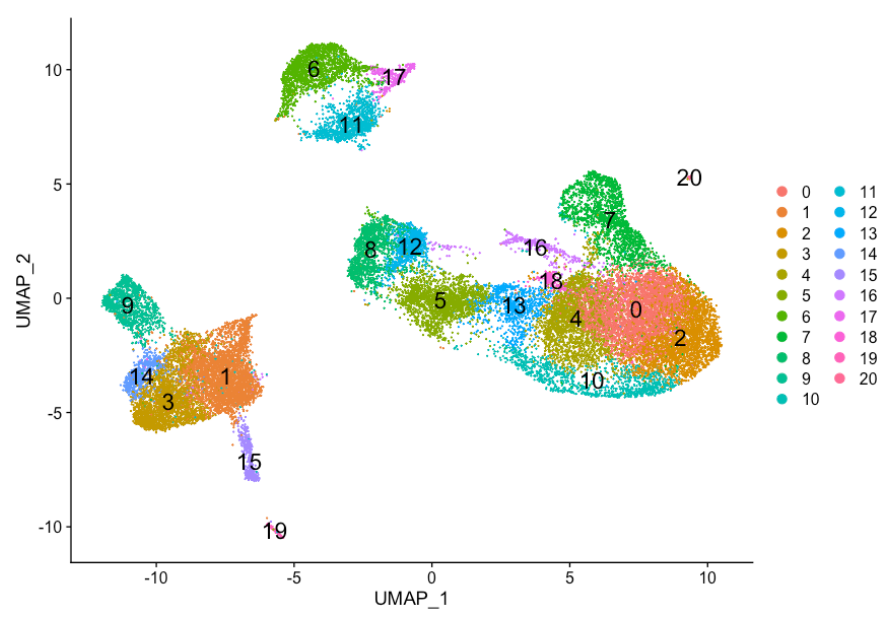
前面我们已经确定了我们想要的簇,我们可以继续进行标记识别,这将使我们能够验证某些簇的身份并帮助推测任何未知簇的身份。 我们之前的聚类分析结果如下: 记住,我们在聚类分析中遇到了以下问题: 我们可以使用 该分析将每个簇与所有其他簇进行比较,并输出差异表达的基因。可用于识别未知簇和提高对假设细胞类型的置信度。 该分析首先寻找在每个条件下差异表达的基因,然后报告在所有条件下在簇中保守的那些基因。这些基因可以帮助确定簇的身份。适用于多个条件以识别跨条件保守的细胞类型标记。 该分析探讨了特定簇之间的差异表达基因。用于确定上述分析中似乎代表相同细胞类型(即具有相似标记)的簇之间基因表达的差异。 在评估单个样品时,通常建议使用这种类型的分析。使用 注意:默认为 Wilcoxon 秩和检验,但还有其他可用选项。 您可以使用这些参数的任意组合,具体取决于您想要的严格程度。此外,默认情况下,此函数将返回给您显示阳性和阴性表达变化的基因。通常,我们添加一个参数 由于我们的数据集中有代表不同条件的样本,我们最好的选择是找到保守的标记。此方法在内部按样本组/条件分离细胞,然后针对所有其他簇(或第二个簇,如果指定)对单个指定簇执行差异基因表达测试。计算每个条件的基因水平 p 值,然后使用 在我们开始我们的标记识别之前,我们将明确设置我们的默认分析,我们想要使用标准化数据,而不是簇数据。 您将认识到我们之前为 对于我们的分析,相当宽松,仅使用大于 0.25 的对数倍数变化阈值。我们还将指定只返回每个簇的正标记。 看看它是如何在一个簇上工作的: 在查看输出时,我们建议寻找 添加带有基因注释信息的列会很有帮助。为此,我们将使用下面提供的代码加载位于您的数据文件夹中的注释文件: 首先,我们将带有基因标识符的行名转换为自己的列。然后我们将这个注释文件与 函数 现在我们已经创建了这个函数,我们可以将它用作适当映射函数的参数。我们希望 现在,让我们尝试使用此函数来查找未识别细胞类型簇的保守标记:簇 7 和簇 20。 我们想使用这些基因列表来查看我们可以识别这些簇识别的细胞类型。让我们看看每个簇的Top基因。我们可以通过两组的平均倍数变化来查看前 10 个标记,以便快速浏览每个簇: 我们看到簇 7 出现了很多热休克和 DNA 损伤基因。基于这些标记,这些很可能是压力或垂死的细胞。然而,如果我们更详细地探索这些细胞的质量指标(即覆盖在簇上的 对于簇 20,富集的基因似乎没有一个共同关系。我们查看 因此簇20可能代表肥大细胞。肥大细胞是免疫系统的重要细胞,属于造血谱系。研究已经确定肥大细胞特征显着富含丝氨酸蛋白酶,例如 TPSAB1 和 TPSB2,这两种蛋白酶都出现在我们的保守标记列表中。另一个不是丝氨酸蛋白酶,而是已知的肥大细胞特异性基因并出现在我们列表中的基因是 FCER1A(编码 IgE 受体的一个亚基)。此外,我们看到 GATA1 和 GATA2 出现在我们的列表中,它们不是肥大细胞标记基因,而是在肥大细胞中大量表达,并且是调节各种肥大细胞特异性基因的已知转录因子。 为了更好地了解簇 20 的细胞类型身份,我们可以使用 我们还可以使用小提琴图来探索特定标记的表达范围: 这些结果和图表可以帮助我们确定这些簇的身份,或者验证我们在之前探索预期细胞类型的规范标记后假设的身份。 关于分析的最后一组问题涉及对应于相同细胞类型的簇是否具有生物学意义的差异。有时返回的标记列表不能充分分离某些簇。例如,我们之前已将 0、2、4、10 和 18 号簇确定为 CD4+ T 细胞,但这些细胞簇之间是否存在生物学相关差异?我们可以使用 我们可以尝试所有比较组合,但我们将从簇 2 与所有其他 CD4+ T 细胞簇开始: 在这些Top基因中,CREM 基因作为激活的标志物脱颖而出。我们知道另一个激活标志物是 CD69,而幼或记忆细胞的标志物包括 SELL 和 CCR7 基因。有趣的是,SELL 基因也位居榜首。让我们使用这些新的细胞状态标记来直观地探索激活状态: 由于初始状态和激活状态的标记都显示在标记列表中,因此可视化表达很有帮助。根据这些图,集群 0 和 2 似乎可靠地是幼稚 T 细胞。然而,对于激活的 T 细胞,很难说。我们可以说簇 4 和 18 是活化的 T 细胞,但 CD69 的表达不如 CREM 明显。我们将标记幼稚细胞并将剩余的簇标记为 CD4+ T 细胞。 现在获取所有这些信息,我们可以推测不同簇的细胞类型并绘制带有细胞类型标签的细胞。 然后我们可以将簇的身份重新分配给这些细胞类型: 如果我们想去除潜在的压力细胞,我们可以使用 现在我们已经定义了簇和每个簇的标记,我们有以下几个不同的选择: 通过实验验证我们识别的细胞类型的标记。 探索细胞类型的子集以发现细胞亚群 > Web[1] 在条件 ctrl 和 stim 之间执行差异表达分析 如果试图确定细胞类型或细胞状态之间的情况,可以进行轨迹分析或谱系追踪: 欢迎Star -> 学习目录 国内链接 -> 学习目录 亚群分析: https://hbctraining.github.io/scRNA-seq_online/lessons/seurat_subclustering.html导读
1. 学习目标
marker
marker识别间进行迭代
2. 目标
3. 挑战
4. 建议
Top markers最可信。在每个条件下识别每个簇的保守标记。
Seurat 探索几种不同类型的标记,来回答这些问题。每个都有自己的优点和缺点:5. 识别每个簇的所有markers
FindAllMarkers() 函数,我们将每个簇与所有其他簇进行比较,以识别潜在的标记基因。每个簇中的细胞被视为重复,本质上是通过一些统计测试进行差异表达分析。
★
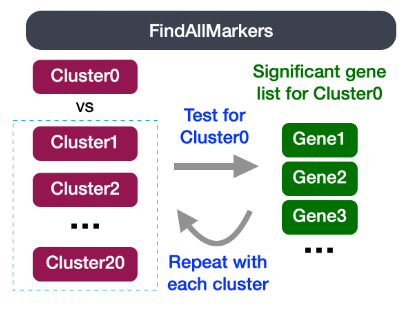
FindAllMarkers() 函数具有三个重要参数,它们提供了确定基因是否为标记的阈值:logfc.threshold:簇中基因平均表达相对于所有其他簇中平均表达的最小log2倍变化。默认值为 0.25。
logfc 不满足阈值,可能会错过那些在感兴趣的簇内的一小部分细胞中表达的细胞标记,但不会在其他簇中表达
min.diff.pct:在簇中表达基因的细胞百分比与在所有其他簇中表达基因的细胞百分比之间的最小百分比差异。
min.pct:仅测试在两个群体中的任何一个中的最小部分细胞中检测到的基因。旨在通过不测试很少表达的基因来加速。默认值为 0.1。
only.pos 来选择只保留积极的变化。为每个簇查找标记的代码如下所示。# 查找每个簇与所有剩余细胞相比的标记,仅报告 positive
markers <- FindAllMarkers(object = seurat_integrated,
only.pos = TRUE,
logfc.threshold = 0.25) 6. 在所有条件下鉴定保守markers
MetaDE R 包中的元分析方法跨组组合。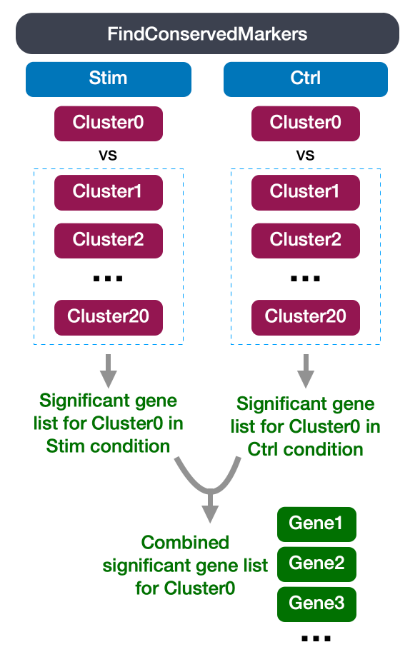
DefaultAssay(seurat_integrated) <- "RNA"FindConservedMarkers() 函数具有以下结构:FindConservedMarkers(seurat_integrated,
ident.1 = cluster,
grouping.var = "sample",
only.pos = TRUE,
min.diff.pct = 0.25,
min.pct = 0.25,
logfc.threshold = 0.25)FindAllMarkers() 函数描述的一些参数;这是因为它在内部使用该函数首先在每个组中查找标记。在这里,我们列出了使用 FindConservedMarkers() 时提供的一些附加参数:ident.1:此函数一次只评估一个簇;在这里,您将指定感兴趣的簇。
grouping.var:元数据中的变量(列标题),它将指定细胞分成组
cluster0_conserved_markers <- FindConservedMarkers(seurat_integrated,
ident.1 = 0,
grouping.var = "sample",
only.pos = TRUE,
logfc.threshold = 0.25)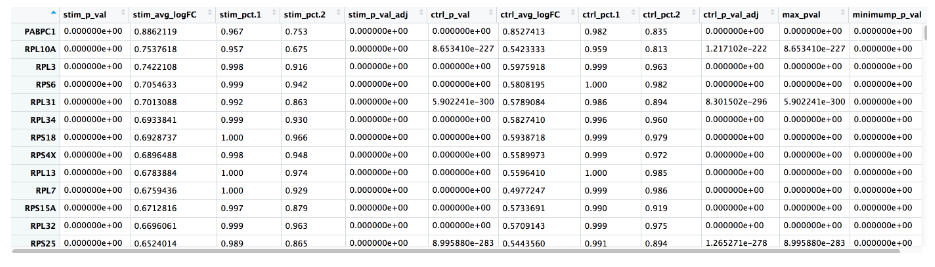
FindConservedMarkers() 函数的输出是一个矩阵,其中包含按我们指定的簇的基因 ID 列出的推定标记的排名列表,以及相关的统计数据。请注意,为每个组(在我们的示例中为 Ctrl 和 Stim)计算相同的统计数据集,最后两列对应于两个组的组合 p 值。我们在下面描述了其中一些列:bonferroni 校正,用于确定显著性
pct.1 和 pct.2 之间表达差异较大且倍数变化较大的标记。例如,如果 pct.1 = 0.90 和 pct.2 = 0.80,它可能不是正确的标记。但是,如果 pct.2 = 0.1 而不是,更大的差异会更有说服力。此外,有趣的是,如果大多数表达标记的细胞都在我感兴趣的簇中,如 pct.1 很低,比如 0.3,它可能不是正确的标记。如上所述,这两个也是运行函数时可能包含的参数。6.1. 添加基因注释
annotations <- read.csv("data/annotation.csv")FindConservedMarkers() 的结果合并:# 将标记与基因描述相结合
cluster0_ann_markers <- cluster0_conserved_markers %>%
rownames_to_column(var="gene") %>%
left_join(y = unique(annotations[, c("gene_name", "description")]),
by = c("gene" = "gene_name"))
View(cluster0_ann_markers)6.2. 在多个样本上运行
FindConservedMarkers() 一次接受一个簇,我们可以运行这个函数的次数与我们簇一样多。但是,这不是很有效。相反,我们将首先创建一个函数来查找包含我们想要包含的所有参数的保守标记。我们还将添加几行代码来修改输出。我们的步骤是:FindConservedMarkers() 函数
rownames_to_column() 函数将行名传输到列
cbind() 函数创建集群
ID 列
# 创建函数以获取任何给定簇的保守标记
get_conserved <- function(cluster){
FindConservedMarkers(seurat_integrated,
ident.1 = cluster,
grouping.var = "sample",
only.pos = TRUE) %>%
rownames_to_column(var = "gene") %>%
left_join(y = unique(annotations[, c("gene_name", "description")]),
by = c("gene" = "gene_name")) %>%
cbind(cluster_id = cluster, .)
}map 系列函数的输出是一个数据帧,每个簇输出由行绑定在一起,我们将使用 map_dfr() 函数。# 示例
map_dfr(inputs_to_function, name_of_function)conserved_markers <- map_dfr(c(7,20), get_conserved)6.3. 评估标记基因
# 每提取个簇前 10 个标记
top10 <- conserved_markers %>%
mutate(avg_fc = (ctrl_avg_log2FC + stim_avg_log2FC) /2) %>%
group_by(cluster_id) %>%
top_n(n = 10,
wt = avg_fc)
# 可视化每个簇的前 10 个标记
View(top10)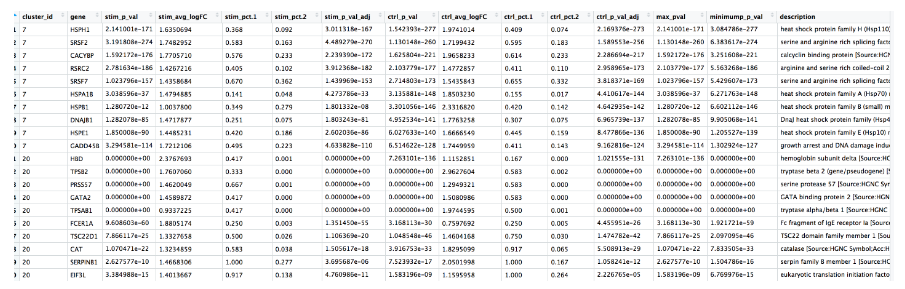
mitoRatio 和 nUMI),我们并没有真正看到支持该论点的数据。如果我们仔细查看标记基因列表,我们还会发现一些 T 细胞相关基因和激活标记。这些可能是激活的(细胞毒性)T细胞。有大量研究支持热休克蛋白与反应性 T 细胞在慢性炎症中诱导抗炎细胞因子的关联。这是一个簇,我们需要对免疫细胞有更深入的了解,才能真正梳理结果并得出最终结论。pct.1 与 pct.2 差异较大的基因,以获得良好的标记基因。例如,我们可能对基因 TPSB2 感兴趣,它显示簇中的大部分细胞表达该基因,但其他簇中表达该基因的细胞很少。6.4. 可视化marker基因
FeaturePlot() 函数按簇探索不同已识别标记的表达。# 绘制簇 20 的标记基因表达
FeaturePlot(object = seurat_integrated,
features = c("TPSAB1", "TPSB2", "FCER1A", "GATA1", "GATA2"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE,
repel = TRUE)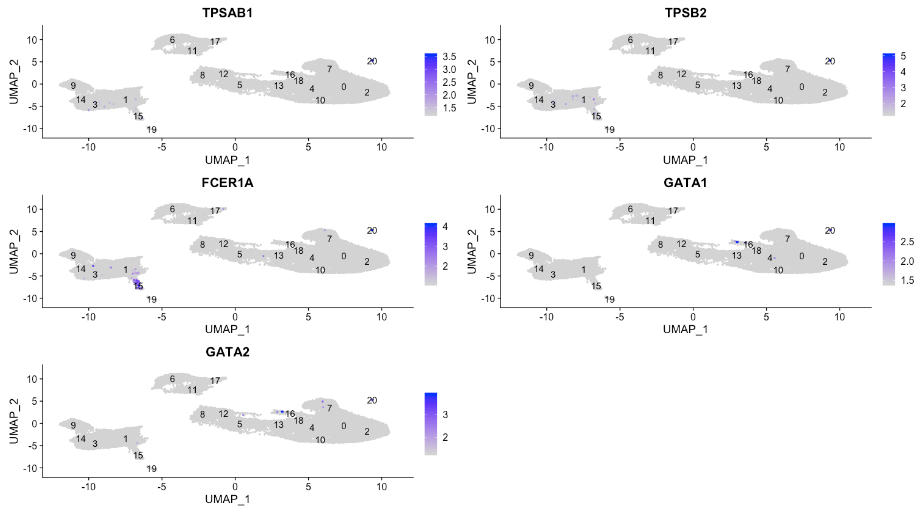
# 小提琴图 - cluster 20
VlnPlot(object = seurat_integrated,
features = c("TPSAB1", "TPSB2", "FCER1A", "GATA1", "GATA2"))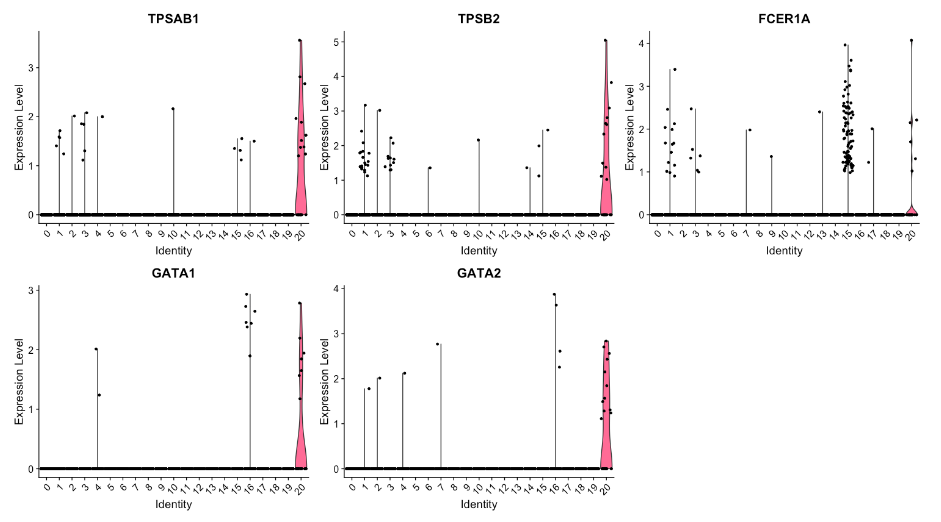
7. 识别每个簇的markers
FindMarkers() 函数来确定在两个特定簇之间差异表达的基因。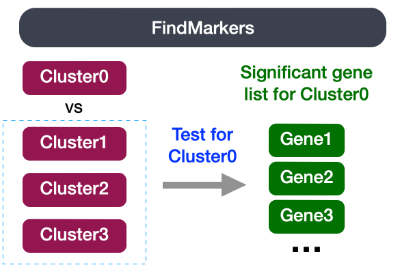
# 确定 CD4+ T 细胞的分化标志物
cd4_tcells <- FindMarkers(seurat_integrated,
ident.1 = 2,
ident.2 = c(0,4,10,18))
# 将基因符号添加到 DE 表
cd4_tcells <- cd4_tcells %>%
rownames_to_column(var = "gene") %>%
left_join(y = unique(annotations[, c("gene_name", "description")]),
by = c("gene" = "gene_name"))
# 重新排序列并按 padj 排序
cd4_tcells <- cd4_tcells[, c(1, 3:5,2,6:7)]
cd4_tcells <- cd4_tcells %>%
dplyr::arrange(p_val_adj)
# 查看数据
View(cd4_tcells)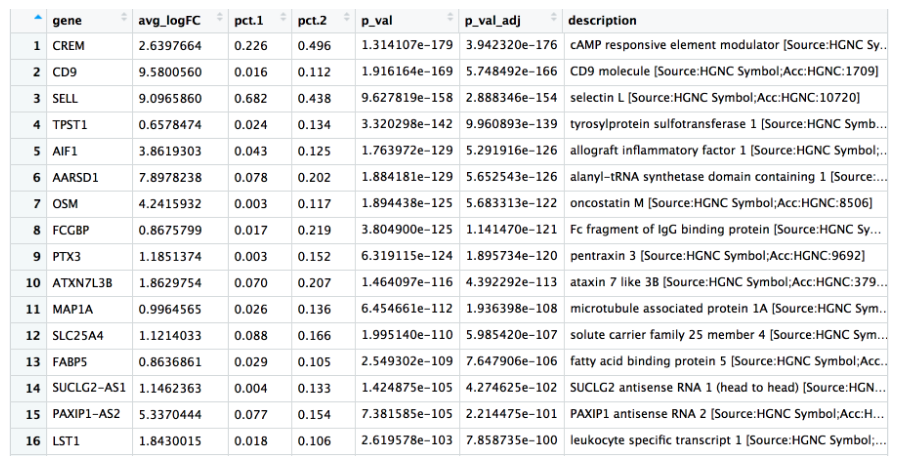
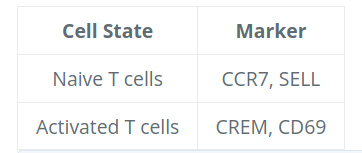
# 绘制激活和记忆 T 细胞的基因标记
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("CREM", "CD69", "CCR7", "SELL"),
label = TRUE,
order = TRUE,
min.cutoff = 'q10',
repel = TRUE
)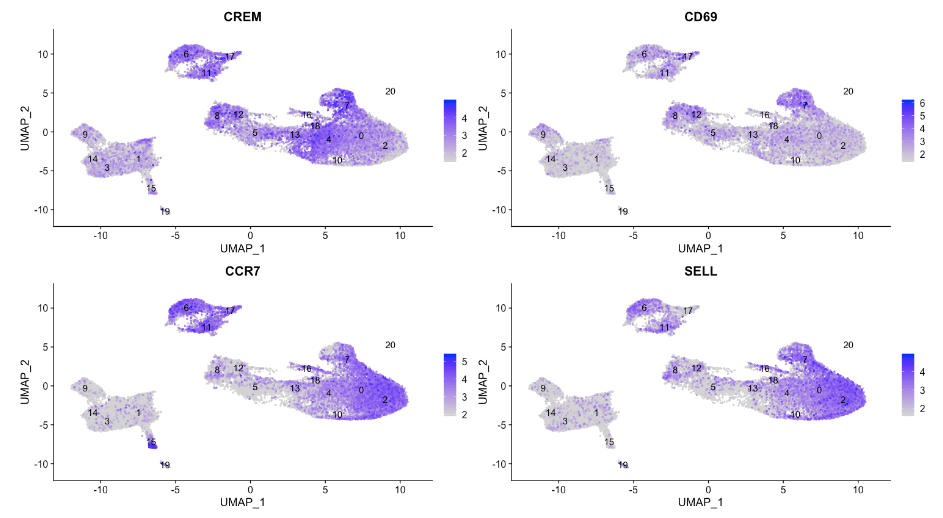
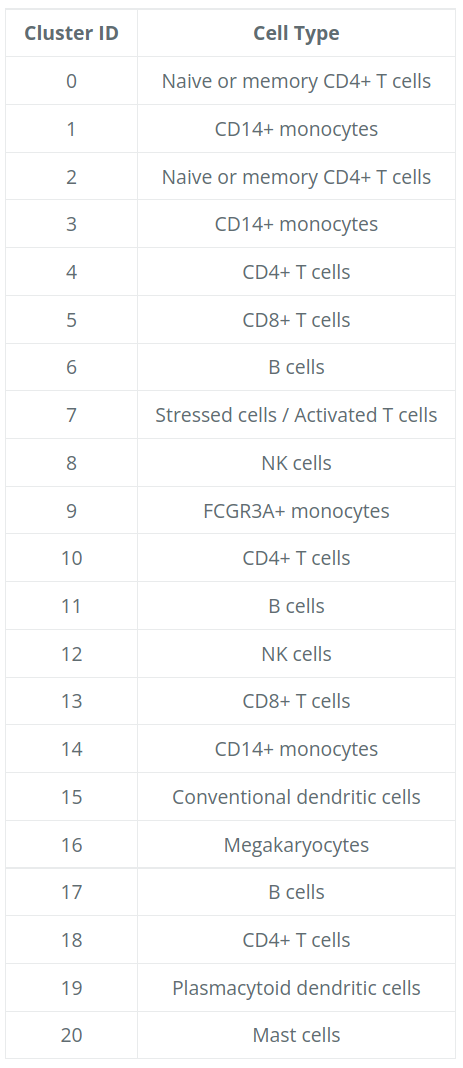
# 重命名所有身份
seurat_integrated <- RenameIdents(object = seurat_integrated,
"0" = "Naive or memory CD4+ T cells",
"1" = "CD14+ monocytes",
"2" = "Naive or memory CD4+ T cells",
"3" = "CD14+ monocytes",
"4" = "CD4+ T cells",
"5" = "CD8+ T cells",
"6" = "B cells",
"7" = "Stressed cells / Activated T cells",
"8" = "NK cells",
"9" = "FCGR3A+ monocytes",
"10" = "CD4+ T cells",
"11" = "B cells",
"12" = "NK cells",
"13" = "CD8+ T cells",
"14" = "CD14+ monocytes",
"15" = "Conventional dendritic cells",
"16" = "Megakaryocytes",
"17" = "B cells",
"18" = "CD4+ T cells",
"19" = "Plasmacytoid dendritic cells",
"20" = "Mast cells")
# UMAP 可视化
DimPlot(object = seurat_integrated,
reduction = "umap",
label = TRUE,
label.size = 3,
repel = TRUE)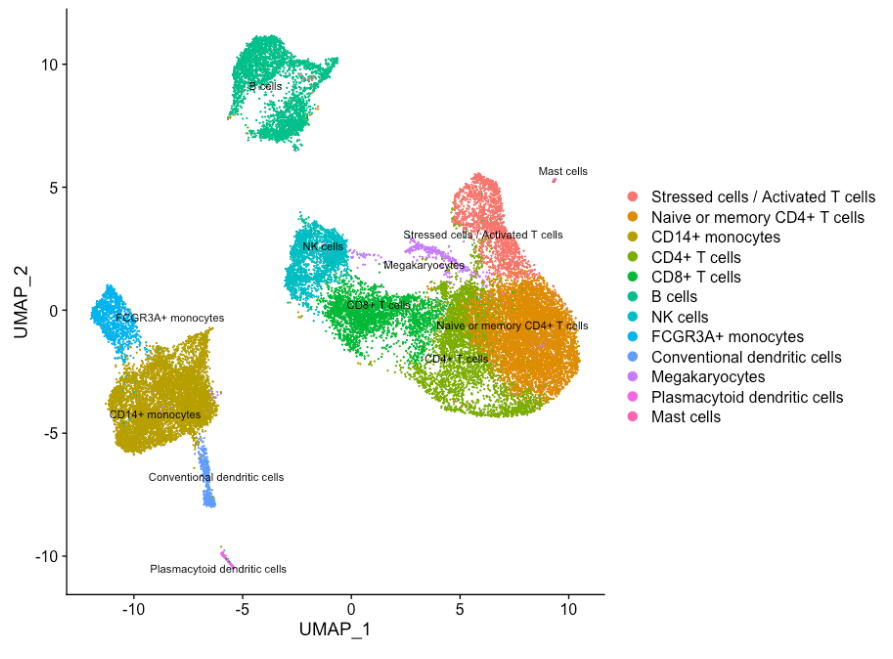
subset() 函数:# 去除垂死的细胞
seurat_subset_labeled <- subset(seurat_integrated,
idents = "Stressed cells / Activated T cells", invert = TRUE)
# 重新可视化簇
DimPlot(object = seurat_subset_labeled,
reduction = "umap",
label = TRUE,
label.size = 3,
repel = TRUE)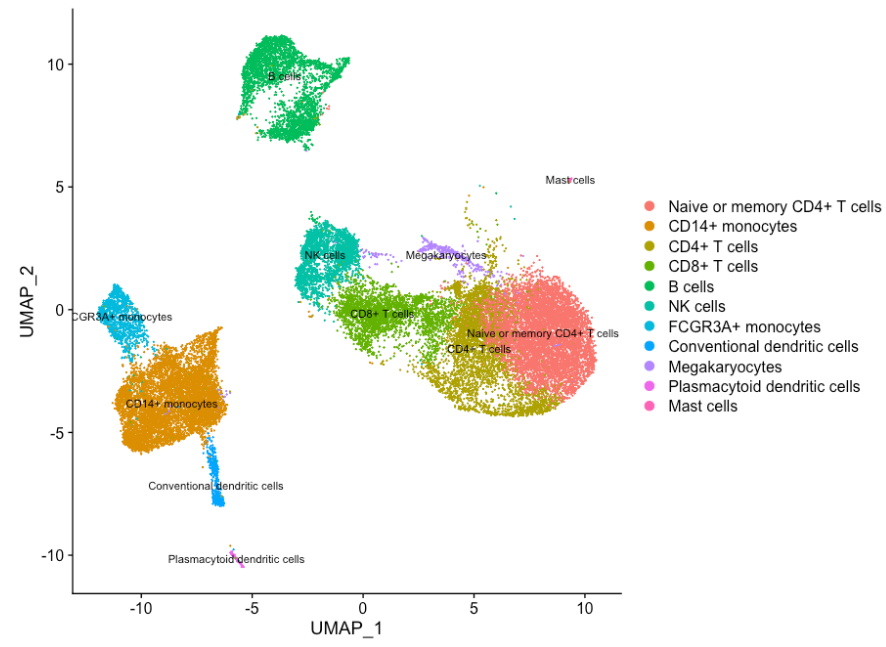
参考资料
本文由 mdnice 多平台发布
最后
以上就是爱撒娇鱼最近收集整理的关于单细胞分析:marker鉴定(11)的全部内容,更多相关单细胞分析内容请搜索靠谱客的其他文章。








发表评论 取消回复