更多内容请关注个人公众号---KS科研分享与服务---
接上节(跟着Cell学单细胞转录组分析(三):单细胞转录组数据质控(QC)及合并去除批次效应)。
数据合并之后,就需要跑标准的Seurat分析流程了。在《cell》文章中,作者还计算了细胞周期评分,因为我们收集到的细胞可能处于不同的分裂时期,所以看周期是很有必要的,尤其是针对具体的研究目的。在示例数据中,可以看到,各个样品细胞周期基本一致。
s.genes <- cc.genes$s.genes
g2m.genes <- cc.genes$g2m.genes
scedata <- CellCycleScoring(scedata,
s.features = s.genes,
g2m.features = g2m.genes,
set.ident = TRUE)
VlnPlot(scedata,features = c("S.Score","G2M.Score"),group.by = "orig.ident")
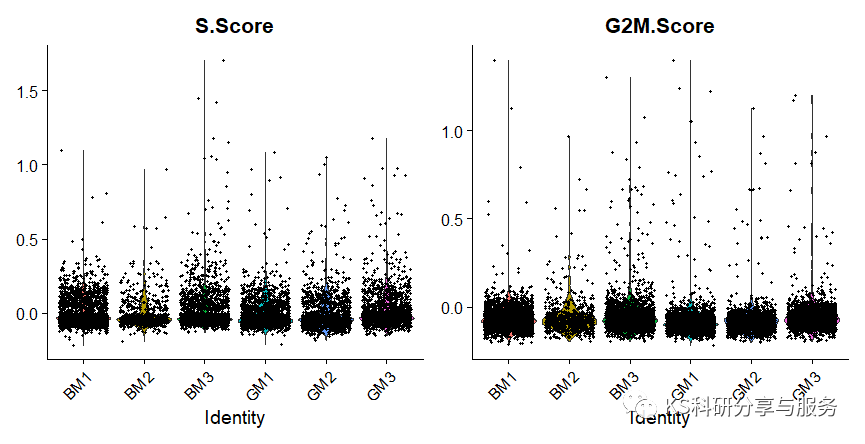
图片
之后对数据进行缩放,缩放的参数vars.to.regress按照自己的目的决定,一般选择percent.mt,nFeature。然后就是确定降维的PC数了,具体选择多少比较合适,这个需要不断的尝试,没有标准,达到自己理想的效果即可。
# 标准流程
scedata <- ScaleData(scedata, vars.to.regress = c("S.Score", "G2M.Score"), verbose = FALSE)
scedata <- RunPCA(scedata, npcs = 50, verbose = FALSE)
scedata <- FindNeighbors(scedata, reduction = "pca", dims = 1:50)
scedata <- FindClusters(scedata,
resolution = seq(from = 0.1,
to = 1.0,
by = 0.1))
scedata <- RunUMAP(scedata, reduction = "pca", dims = 1:50)
最后,决定细胞聚类群的还有一个因素,那就是FindClusters函数中的resolution 这个参数,这里我们直接跑联系的多个resolution,用clustree函数查看。这个参数也是需要调整。
library(clustree)
clustree(scedata)
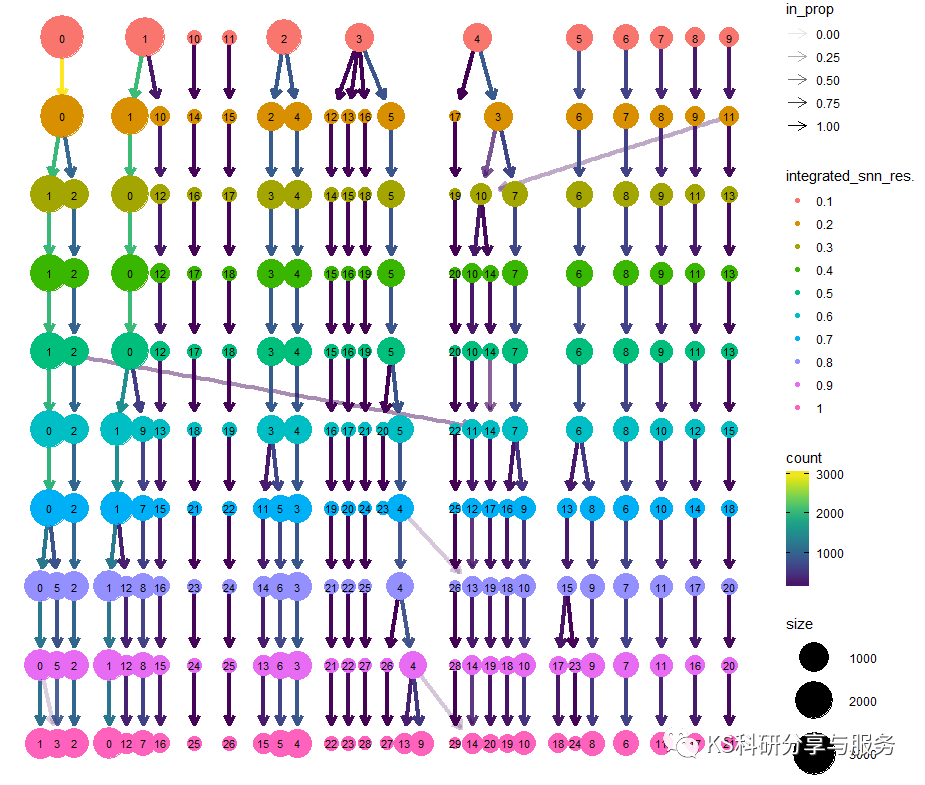
图片
然后就可以对UMAP降维进行可视化了!
DimPlot(scedata)
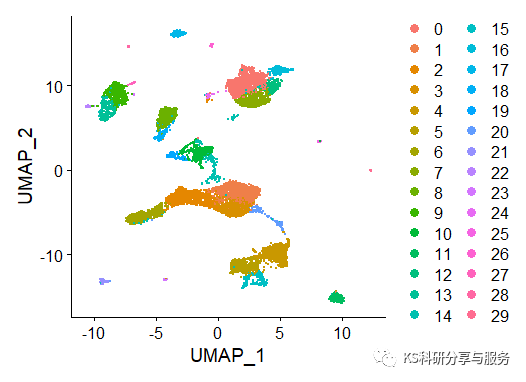
图片
选择好合适的细胞分群,将其设置为active.ident,方便后续可视化。
Idents(scedata) <- "integrated_snn_res.1"
scedata$seurat_clusters <- scedata@active.ident
处理所有细胞的UMAP聚类,查看下细胞群在不同样品中的分布情况。
DimPlot(scedata,label = T,split.by = "orig.ident",ncol = 3)
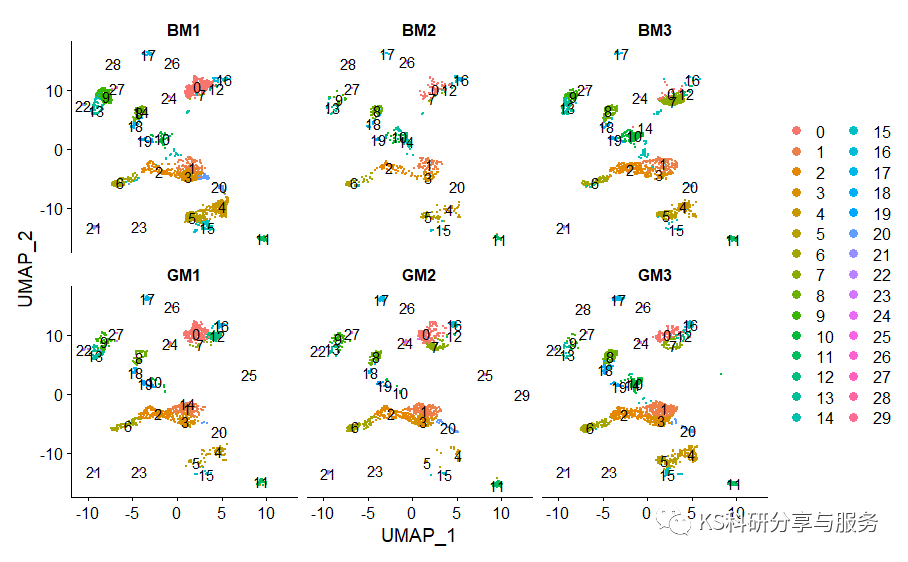
图片
完成上述内容,整个单细胞转录组的工作已经完整了1/3了,因为这些都是后续分析的基础。下节探讨下marker基因的筛选和细胞群鉴定!
最后
以上就是机智水池最近收集整理的关于跟着Cell学单细胞转录组分析(四):单细胞转录组测序UMAP降维聚类的全部内容,更多相关跟着Cell学单细胞转录组分析(四)内容请搜索靠谱客的其他文章。
本图文内容来源于网友提供,作为学习参考使用,或来自网络收集整理,版权属于原作者所有。







发表评论 取消回复