单细胞选取数据
1.常用的几种数据含义
Assays
默认情况下,我们是对Seurat中的RNA的Assay进行操作。最初开始的数据只有一个RNA的assay,数据在进行变换SCT,或者整合单样本数据intergration,或去除污染SoupX,或融合velocity;都会将处理后的结果数据存放到新的Assay中。
可以通过@active.assay查看当前默认的assay,通过DefaultAssay()更改当前的默认assay。
counts为raw原始数据,我们开始对assay中的RNA数据中的counts进行处理,对稀疏矩阵进行归一化处理后的结果存储在data, 接着用ScaleData()处理后的数据存储在scale.data中。
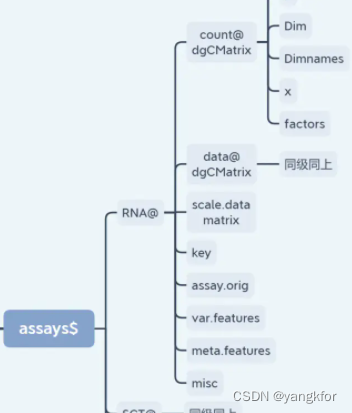
三种数据之间的结构
counts:存储原始数据,是稀疏矩阵
data:存储logNormalize() 规范化的data。总表达式对每个单元格的要素表达式度量进行标准化,将其乘以比例因子(默认为10,000),并对结果进行对数转换
scale.data: 存储 ScaleData()缩放后的data,此步骤需要时间久。
每个数据都是一个单独的数据框,列名都是一样的,但是存放的数据不一样。
调用方法: head(pbmc@assays$RNA@data) :调用raw数据
meta.data
元数据,对每个细胞的描述。一般的meta.data包括orig.ident, nCount_RNA, nFeature_RNA, 以及计算后的percent.mt,RNA_snn_res.0.5等;后期的线粒体过滤、细胞个数是通过该表进行操作的。
调用方式:pbmc$percent.mt或pbmc[['percent.mt']]:查看percent.mt比例,一个是data.frame;一个是向量。
reduction
降维后的每个细胞的坐标信息,包括pca,tsne,umap等;将integrated作为默认assay进行PCAUMAP等分析。
2. 对象操作
① 通过结构图上的@,$符号依次取
② 两个中括号操作,pbmc[[ ]]
教程中,pbmc[[‘percent.MT’]]向meta.data添加 percent.MT 这一列。
pbmc[[]],中括号取的是上面结构图中的二级数据名称
@,
符
号
依
次
取
,
是
两
个
符
号
交
替
进
行
,
先
@
在
符号依次取,是两个符号交替进行,先@在
符号依次取,是两个符号交替进行,先@在,如pbmc@assays$RNA@data;此处没有$之后接向量的含义,跟R选取数据不同。
差异
针对assay或者resolution而言,两种方式得到的结果不存在差别
pbmc[['RNA']] 或者 pbmc@assays$RNA
pbmc[['pca']] 或者pbmc@reductions$pca
针对meta.data的分析内容存在差别
pbmc[['nCount_RNA']] #取出来是所有细胞的nCount_RNA,是一个数据框
pbmc@meta.data$nCount_RNA #取出来的是单独nCount_RNA一列,是向量
最后
以上就是传统树叶最近收集整理的关于单细胞选取数据的全部内容,更多相关单细胞选取数据内容请搜索靠谱客的其他文章。







发表评论 取消回复