分享是一种态度

作者 | 周运来
男,
一个长大了才会遇到的帅哥,
稳健,潇洒,大方,靠谱。
一段生信缘,一棵技能树。
生信技能树核心成员,单细胞天地特约撰稿人,简书创作者,单细胞数据科学家。
前情回顾
Seurat 4.0 ||您的单细胞数据分析工具箱上新啦
Seurat 4.0 ||单细胞多模态数据整合算法WNN
Seurat 4.0 || 分析scRNA和膜蛋白数据
Seurat 4.0 || WNN整合scRNA和scATAC数据
正文
细胞不只是RNA的盒子,而是中心法则的反应器。随着单细胞技术的成熟,同一细胞不同模态的数据也被同时测量出来。在scRNA时代,细胞类型的鉴定往往依靠RNA表达谱或者几个标记基因。多模态分析提供了更接近细胞真实状态的数据,也为细胞类型的鉴定提供了新的可能。Seurat 升级到4.0时,同时提供了基于RNA和膜蛋白的PBMC参考数据集,这可以说进一步解决了PBMC细胞类型(状态)的鉴定难题。PBMC的scRNA数据应用这个数据集和算法,基本可以得到很好的注释。Seurat官网的教程介绍了在Seurat中将查询数据集(query )映射到参考数据集(references )上的过程。在本例中,我们将10X Genomics发布的首批2700 PBMC的scRNA-seq数据集映射到我们最近描述的包含228(224?)抗体的162,000 PBMC的CITE-seq参考数据集上。我们选择此示例是为了演示由参考数据集指导的监督分析,如何有助于找出在非监督分析中难以找到的细胞状态。在第二个示例中,我们将演示如何将不同个体的人类BMNC的人类细胞图谱数据集映射到参考数据上。
当然,Seurat提供了Azimuth,一个利用高质量参考数据集快速映射新的scRNA-seq数据集(查询)的界面工具。例如,您可以将人类PBMC的任何scRNA-seq数据集映射到我们的references上,从而自动化可视化、聚类注释和差异表达的过程。Azimuth可以在Seurat内运行,也可以使用不需要安装或编程经验的独立web应用程序运行。
- Web app: Automated mapping, visualization, and annotation of scRNA-seq datasets from human PBMC
我们这里展示的当然是如何在R里面运行了呀。
我们前面 单细胞转录组数据分析||Seurat新版教程: Integration and Label Transfer演示了如何使用参考数据映射方法在查询数据集中注释细胞标签。在Seurat v4中,我们优化了集成任务(包括参考数据集映射)的速度和内存需求,还包括将查询细胞投影到以前计算的UMAP空间中。
在这里,我们演示如何使用先前建立的参考数据来注释一个待查询的scRNA数据:
- 根据一组参考数据定义细胞状态来注释每个待注释的细胞
- 将每个查询数据集投射到以前计算的UMAP空间中
- 根据CITE-seq 参考数据集预测中表面蛋白表达水平
要进行这个分析,请安装Seurat v4,在我们的github页面上有beta版本。此外,还需要安装最新版本的uwot包和SeuratDisk。
remotes::install_github("satijalab/seurat", ref = "release/4.0.0")
remotes::install_github("jlmelville/uwot")
remotes::install_github("mojaveazure/seurat-disk")
library(Seurat)
library(SeuratDisk)
library(ggplot2)
library(patchwork)
reference "../data/pbmc_multimodal.h5seurat")
Validating h5Seurat file
Initializing ADT with data
Adding counts for ADT
Initializing SCT with data
Adding counts for SCT
Adding variable feature information for SCT
Adding miscellaneous information for SCT
Adding reduction apca
Adding cell embeddings for apca
Adding feature loadings for apca
Adding miscellaneous information for apca
Adding reduction aumap
Adding cell embeddings for aumap
Adding miscellaneous information for aumap
Adding reduction pca
Adding cell embeddings for pca
Adding feature loadings for pca
Adding miscellaneous information for pca
Adding reduction spca
Adding cell embeddings for spca
Adding feature loadings for spca
Adding miscellaneous information for spca
Adding reduction umap
Adding cell embeddings for umap
Adding miscellaneous information for umap
Adding reduction wnn.umap
Adding cell embeddings for wnn.umap
Adding miscellaneous information for wnn.umap
Adding graph wknn
Adding graph wsnn
Adding command information
Adding cell-level metadata
Adding miscellaneous information
Adding tool-specific results
可以看到这个参考数据集做了基本的计算,并以h5Seurat文件的形式存储,这种格式允许在磁盘上存储多模式Seurat对象。
reference
An object of class Seurat
20953 features across 161764 samples within 2 assays
Active assay: SCT (20729 features, 5000 variable features)
1 other assay present: ADT
6 dimensional reductions calculated: apca, aumap, pca, spca, umap, wnn.umap
reference@commands
list()# 作者没有保留其计算过程
DimPlot(object = reference, reduction = "wnn.umap", group.by = "celltype.l2", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
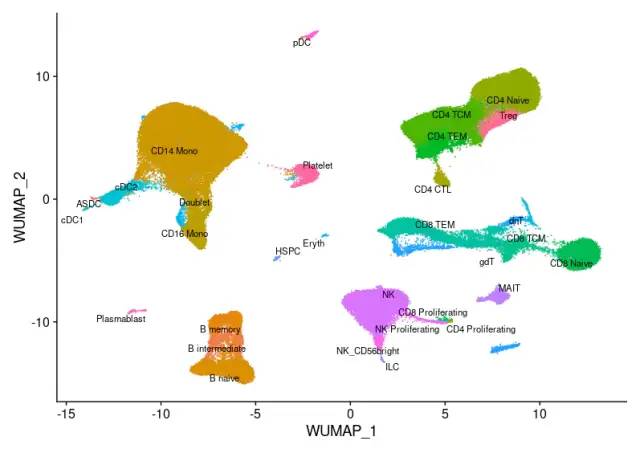
为了演示到这个多模态参考的映射,我们将使用由10x Genomics产生的2700个PBMCs的数据集,并通过SeuratData包调取。
library(SeuratData)
#InstallData('pbmc3k')# 我之前安装过了
参考数据集是使用SCTransform规范化的,因此我们在这里使用相同的方法规范化查询数据集。
pbmc3k
然后我们在参考集和查询集之间找锚点(FindTransferAnchors)。我们在这个例子中使用了预先计算的监督PCA (spca)转换。我们建议对CITE-seq数据集使用监督PCA(supervised PCA ),并在本示例上演示如何计算这种转换。但是,您也可以使用标准的PCA转换。出于探索性分析的考虑,在不能抉择的时候,就都试一试。
anchors reference = reference,
query = pbmc3k,
normalization.method = "SCT",
reference.reduction = "spca",
dims = 1:50
)
Projecting cell embeddings
Finding neighborhoods
Finding anchors
Found 6768 anchors
anchors
An AnchorSet object containing 6768 anchors between the reference and query Seurat objects.
This can be used as input to TransferData.
然后我们用MapQuery函数将参考集中的细胞类型标签和蛋白质数据转移到查询集中。此外,我们将查询数据投影到参考集的UMAP结构上。
?MapQuery
pbmc3k anchorset = anchors,
query = pbmc3k,
reference = reference,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT"
),
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
head(pbmc3k@meta.data)
orig.ident nCount_RNA nFeature_RNA seurat_annotations nCount_SCT nFeature_SCT predicted.celltype.l1.score
AAACATACAACCAC pbmc3k 2419 779 Memory CD4 T 2292 779 0.9454906
AAACATTGAGCTAC pbmc3k 4903 1352 B 2633 1089 1.0000000
AAACATTGATCAGC pbmc3k 3147 1129 Memory CD4 T 2485 1126 1.0000000
AAACCGTGCTTCCG pbmc3k 2639 960 CD14+ Mono 2364 958 1.0000000
AAACCGTGTATGCG pbmc3k 980 521 NK 1980 560 1.0000000
AAACGCACTGGTAC pbmc3k 2163 781 Memory CD4 T 2168 781 0.9768638
predicted.celltype.l1 predicted.celltype.l2.score predicted.celltype.l2
AAACATACAACCAC CD8 T 0.7605497 CD8 TCM
AAACATTGAGCTAC B 0.5192524 B intermediate
AAACATTGATCAGC CD4 T 0.8240204 CD4 TCM
AAACCGTGCTTCCG Mono 0.9691920 CD14 Mono
AAACCGTGTATGCG NK 1.0000000 NK
AAACGCACTGGTAC CD4 T 0.7975231 Treg
head(reference@meta.data)
nCount_ADT nFeature_ADT nCount_RNA nFeature_RNA orig.ident lane donor time celltype.l1 celltype.l2
L1_AAACCCAAGAAACTCA 7535 217 10823 2915 P2_7 L1 P2 7 Mono CD14 Mono
L1_AAACCCAAGACATACA 6013 209 5864 1617 P1_7 L1 P1 7 CD4 T CD4 TCM
L1_AAACCCACAACTGGTT 6620 213 5067 1381 P4_3 L1 P4 3 CD8 T CD8 Naive
L1_AAACCCACACGTACTA 3567 202 4786 1890 P3_7 L1 P3 7 NK NK
L1_AAACCCACAGCATACT 6402 215 6505 1621 P4_7 L1 P4 7 CD8 T CD8 Naive
L1_AAACCCACATCAGTCA 5297 212 4332 1633 P3_3 L1 P3 3 CD8 T CD8 TEM
celltype.l3 Phase
L1_AAACCCAAGAAACTCA CD14 Mono G1
L1_AAACCCAAGACATACA CD4 TCM_1 G1
L1_AAACCCACAACTGGTT CD8 Naive S
L1_AAACCCACACGTACTA NK_2 G1
L1_AAACCCACAGCATACT CD8 Naive G1
L1_AAACCCACATCAGTCA CD8 TEM_1 G1
MapQuery是一个围绕三个函数的包装器:TransferData、IntegrateEmbeddings和ProjectUMAP。TransferData用于传输细胞类型标签和输入ADT值。IntegrateEmbeddings和ProjectUMAP用于将查询数据投影到参考集的UMAP结构上。下面是与中间函数等价的代码:
pbmc3k anchorset = anchors,
reference = reference,
query = pbmc3k,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT")
)
pbmc3k anchorset = anchors,
reference = reference,
query = pbmc3k,
new.reduction.name = "ref.spca"
)
pbmc3k query = pbmc3k,
query.reduction = "ref.spca",
reference = reference,
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
我们现在可以可视化2700个查询数据集。它们已经被投射到由参考集定义的UMAP空间中,并且每个都收到了两个粒度级别(级别1和级别2)的注释。
p1 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l1", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
p2 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l2", label = TRUE, label.size = 3 ,repel = TRUE) + NoLegend()
p1 + p2
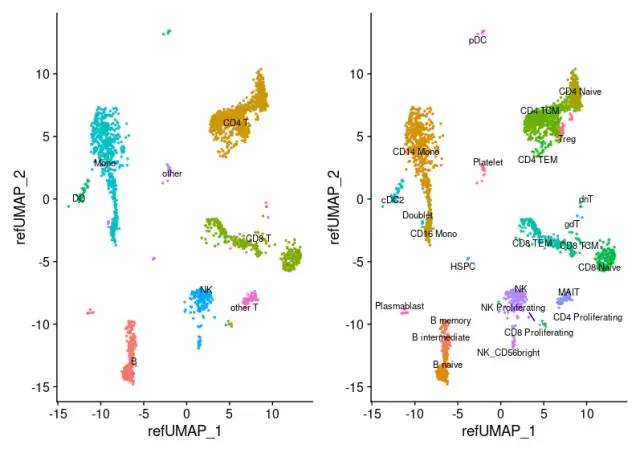
这里我们一定要用桑基图看不同注释结果的拟合程度啊。
library(Seurat)
library(ggforce)
pbmc3k@meta.data %>% na.omit() %>%
gather_set_data(c(4,8,10)) %>%
ggplot(aes(x, id = id, split = y, value = 1)) +
geom_parallel_sets(aes(fill = seurat_annotations ), show.legend = FALSE, alpha = 0.3) +
geom_parallel_sets_axes(axis.width = 0.1, color = "red", fill = "white") +
geom_parallel_sets_labels(angle = 0) +
theme_no_axes()
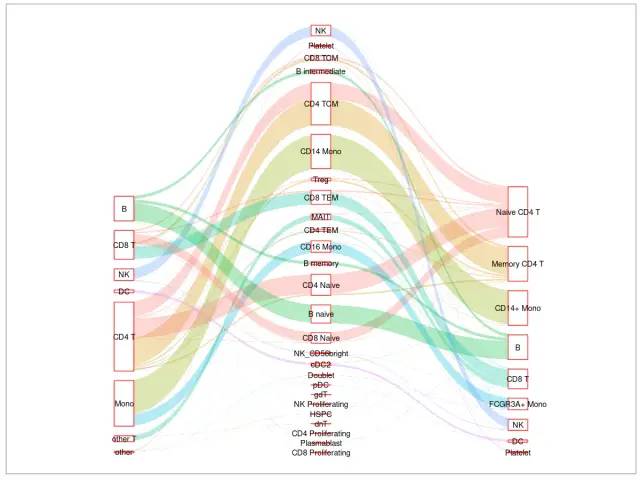
参考映射数据集帮助我们识别以前在查询数据集的非监督分析中混合的细胞类型。一些例子包括浆细胞样树突状细胞(pDC),造血干细胞和祖细胞(HSPC),调节性T细胞(Treg), CD8幼稚T细胞,细胞,CD56+ NK细胞,记忆,和幼稚B细胞,血浆再生细胞(plasmablasts)。
每个预测都被赋一个0到1之间的分数。
head(pbmc3k@meta.data)
orig.ident nCount_RNA nFeature_RNA seurat_annotations nCount_SCT
AAACATACAACCAC pbmc3k 2419 779 Memory CD4 T 2292
AAACATTGAGCTAC pbmc3k 4903 1352 B 2633
AAACATTGATCAGC pbmc3k 3147 1129 Memory CD4 T 2485
AAACCGTGCTTCCG pbmc3k 2639 960 CD14+ Mono 2364
AAACCGTGTATGCG pbmc3k 980 521 NK 1980
AAACGCACTGGTAC pbmc3k 2163 781 Memory CD4 T 2168
nFeature_SCT predicted.celltype.l1.score predicted.celltype.l1
AAACATACAACCAC 779 0.9454906 CD8 T
AAACATTGAGCTAC 1089 1.0000000 B
AAACATTGATCAGC 1126 1.0000000 CD4 T
AAACCGTGCTTCCG 958 1.0000000 Mono
AAACCGTGTATGCG 560 1.0000000 NK
AAACGCACTGGTAC 781 0.9768638 CD4 T
predicted.celltype.l2.score predicted.celltype.l2
AAACATACAACCAC 0.7605497 CD8 TCM
AAACATTGAGCTAC 0.5192524 B intermediate
AAACATTGATCAGC 0.8240204 CD4 TCM
AAACCGTGCTTCCG 0.9691920 CD14 Mono
AAACCGTGTATGCG 1.0000000 NK
AAACGCACTGGTAC 0.7975231 Treg
FeaturePlot(pbmc3k, features = c("pDC", "CD16 Mono", "Treg"), reduction = "ref.umap", cols = c("lightgrey", "darkred"), ncol = 3) & theme(plot.title = element_text(size = 10))
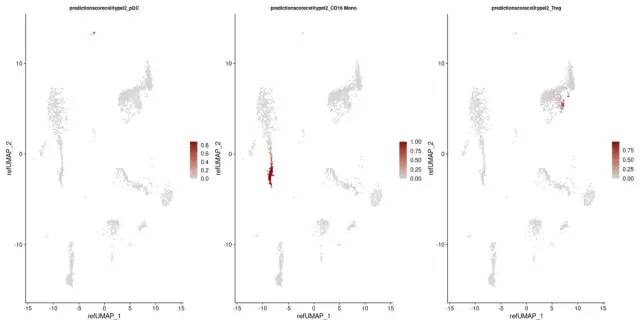
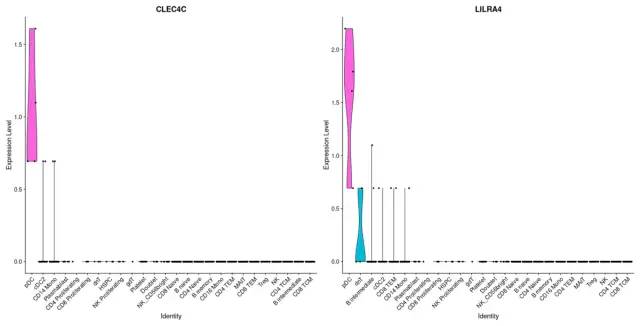
我们可以通过探索典型标记基因的表达来验证我们的预测j结果。例如,CLEC4C和LIRA4已被报道为pDC识别的标记,这与我们的预测一致。同样,如果我们通过差异表达来识别Treg的标记,我们可以识别一组典型标记,包括RTKN2、CTLA4、FOXP3和IL2RA。
Idents(pbmc3k) 'predicted.celltype.l2'
VlnPlot(pbmc3k, features = c("CLEC4C", "LILRA4"), sort = TRUE) + NoLegend()
treg_markers "Treg", only.pos = TRUE, logfc.threshold = 0.1)
print(head(treg_markers))
p_val avg_log2FC pct.1 pct.2 p_val_adj
RTKN2 1.752644e-59 0.3402679 0.216 0.003 2.203424e-55
CTLA4 1.516056e-23 0.1443113 0.108 0.003 1.905986e-19
FOXP3 1.516056e-23 0.1443113 0.108 0.003 1.905986e-19
IL2RA 9.299276e-23 0.3246943 0.216 0.013 1.169105e-18
ZNF485 1.326062e-13 0.1399951 0.108 0.006 1.667126e-09
IL32 4.923202e-13 1.3909210 0.946 0.548 6.189450e-09
最后,我们可以将基于CITE-seq的数据推测的表面蛋白表达水平,这个厉害了啊,我们知道,用我们这个参考数据集我们相当于可以知道224种表面膜蛋白的表达量了(虽然是推测的)。
这224个表面蛋白是:
DefaultAssay(pbmc3k) 'predicted_ADT'
rownames(pbmc3k)
[1] "CD80" "CD86" "CD274" "CD273"
[5] "CD275-1" "CD11b-1" "Galectin-9" "CD270"
[9] "CD252" "CD155" "CD112" "CD47"
[13] "CD70" "CD30" "CD48" "CD40"
[17] "CD154" "CD52" "CD3-1" "CD4-1"
[21] "CD8" "CD56-1" "CD45-1" "CD3-2"
[25] "CD19" "CD11c" "CD34" "CD138-1"
[29] "CD269" "CD90" "CD117" "CD45RA"
[33] "CD123" "CD105" "CD201" "CD194"
[37] "CD4-2" "CD44-1" "CD8a" "CD14"
[41] "CD56-2" "CD25" "CD45RO" "CD279"
[45] "TIGIT" "Rat-IgG2b" "CD20" "CD335"
[49] "CD294" "CD31" "CD44-2" "CD133-1"
[53] "Podoplanin" "CD140a" "CD140b" "Cadherin"
[57] "CD340" "CD146" "CD324" "IgM"
[61] "TCR-1" "CD195" "CD196" "CD185"
[65] "CD103" "CD69" "CD152" "CD223"
[69] "CD27" "CD107a" "CD95" "CD134"
[73] "HLA-DR" "CD1c" "CD11b-2" "CD64"
[77] "CD141" "CD1d" "CD314" "CD66b"
[81] "CD35" "CD57" "CD366" "CD272"
[85] "CD278" "CD275-2" "CD96" "CD39"
[89] "CD178" "CX3CR1" "CD24" "CD21"
[93] "CD79b" "CD66a/c/e" "CD244" "CD235ab"
[97] "Siglec-8" "CD206" "CD169" "CD370"
[101] "XCR1" "Notch-1" "Integrin-7" "CD268"
[105] "CD42b" "CD54" "CD62P" "CD119"
[109] "TCR-2" "Notch-2" "CD68" "Rat-IgG1-1"
[113] "Rat-IgG1-2" "Rag-IgG2c" "CD192" "CD102"
[117] "CD106" "CD122" "CD267" "CD62E"
[121] "CD135" "CD41" "CD137" "CD43"
[125] "CD163" "CD83" "CD357" "CD59"
[129] "CD309" "CD124" "CD13" "CD184"
[133] "CD2" "CD29" "CD303" "CD49b"
[137] "CD61" "CD81" "CD98" "CD177"
[141] "CD55" "IgD" "CD18" "CD28"
[145] "TSLPR" "CD38-1" "CD127" "CD45-2"
[149] "CD15" "CD22" "CD71" "B7-H4"
[153] "CD26-1" "CD193" "CD115" "CD204"
[157] "CD144" "CD301" "CD1a" "CD207"
[161] "CD63" "CD284" "CD304" "CD36"
[165] "CD172a" "CD85g" "CD38-2" "CD243"
[169] "CD72" "MERTK" "Folate" "TIM-4"
[173] "CD171" "CD325" "CD93" "CD200"
[177] "CD338" "C5L2" "CD235a" "CD49a"
[181] "CD49d" "CD73" "CD79a" "CD9"
[185] "TCR-V-7.2" "TCR-V-2" "TCR-V-9" "TCR-V-24-J-18"
[189] "CD354" "CD202b" "CD305" "LOX-1"
[193] "CD203c" "CD133-2" "CD209" "CD110"
[197] "CD337" "CD253" "CD186" "CD226"
[201] "CD205" "CD109" "CD11a/CD18" "CD126"
[205] "CD164" "CD142" "CD307c/FcRL3" "CD307d"
[209] "CD307e" "CD319" "CD138-2" "CD99"
[213] "CLEC12A" "CD16" "CD161" "CCR10"
[217] "CD271" "GP130" "CD199" "CD45RB"
[221] "CD46" "VEGFR-3" "CLEC2" "CD26-2"
DefaultAssay(pbmc3k) 'predicted_ADT'
# see a list of proteins: rownames(pbmc3k)
FeaturePlot(pbmc3k, features = c("CD3-1", "CD45RA", "IgD"), reduction = "ref.umap", cols = c("lightgrey", "darkgreen"), ncol = 3)
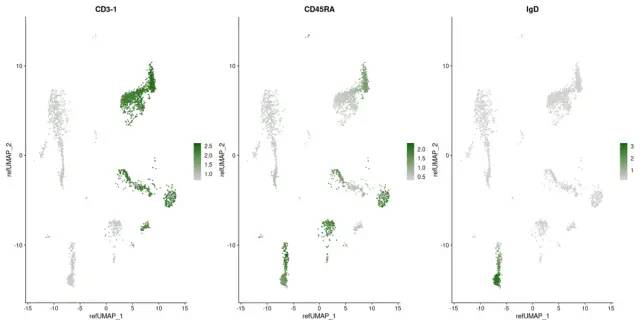
在前面的示例中,我们在映射到参考数据集的UMAP空间可视化了查询数据集。保持一致的可视化可以帮助解释新的数据集。但是,如果查询数据集中存在参考数据集中没有表示的细胞状态,它们将投射到参考数据集中最相似的细胞附近。这是UMAP包所建立的预期行为和功能,但可能会隐藏查询中可能感兴趣的新细胞类型。
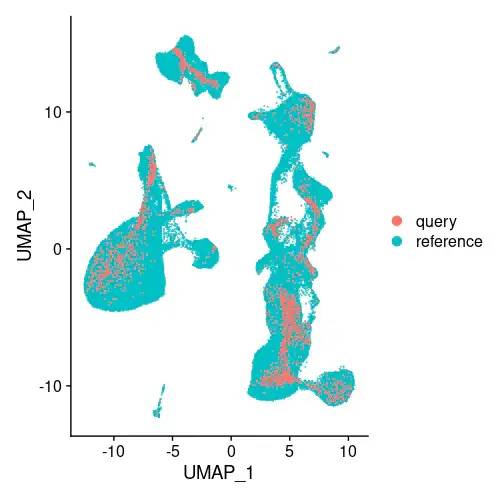
在我们的手稿中,我们绘制了一个查询数据集,包含发展和分化的中性粒细胞,这没有包括在我们的参考数据集中。我们发现,在合并参考集和查询集之后计算一个新的UMAP ('de novo visualization')空间可以帮助识别这些种群,如图所示。在“de novo”可视化中,查询中的唯一细胞状态保持独立。在本例中,2,700 PBMC不包含唯一的细胞状态,但是我们将演示如何计算这种可视化。
我们强调,如果您试图查询的不是PBMC的数据集或者参考集中没有的细胞类型,计算一个“de novo”的可视化是解释数据集的重要步骤。
#merge reference and query
reference$id 'reference'
pbmc3k$id 'query'
refquery refquery[["spca"]] "spca"]], pbmc3k[["ref.spca"]])
refquery 'spca', dims = 1:50)
10:59:22 UMAP embedding parameters a = 0.9922 b = 1.112
10:59:24 Read 164464 rows and found 50 numeric columns
10:59:24 Using Annoy for neighbor search, n_neighbors = 30
10:59:24 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
10:59:46 Writing NN index file to temp file /tmp/RtmppjoJ4T/file5a9164fb867cb
10:59:47 Searching Annoy index using 1 thread, search_k = 3000
11:01:13 Annoy recall = 100%
11:01:14 Commencing smooth kNN distance calibration using 1 thread
11:01:22 Initializing from normalized Laplacian + noise
11:02:34 Commencing optimization for 200 epochs, with 7578986 positive edges
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
11:04:01 Optimization finished
DimPlot(refquery, group.by = 'id')
refquery@commands
$RunUMAP.SCT.spca
Command: RunUMAP(refquery, reduction = "spca", dims = 1:50)
Time: 2020-10-31 11:04:01
dims : 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50
reduction : spca
assay : SCT
slot : data
umap.method : uwot
return.model : FALSE
n.neighbors : 30
n.components : 2
metric : cosine
learning.rate : 1
min.dist : 0.3
spread : 1
set.op.mix.ratio : 1
local.connectivity : 1
repulsion.strength : 1
negative.sample.rate : 5
uwot.sgd : FALSE
seed.use : 42
angular.rp.forest : FALSE
verbose : TRUE
reduction.name : umap
reduction.key : UMAP_
pbmc3k@commands
$SCTransform.RNA
Command: SCTransform(pbmc3k, verbose = FALSE)
Time: 2020-10-31 10:31:19
assay : RNA
new.assay.name : SCT
do.correct.umi : TRUE
ncells : 5000
variable.features.n : 3000
variable.features.rv.th : 1.3
do.scale : FALSE
do.center : TRUE
clip.range : -9.486833 9.486833
conserve.memory : FALSE
return.only.var.genes : TRUE
seed.use : 1448145
verbose : FALSE
References
[1] Automated mapping, visualization, and annotation of scRNA-seq datasets from human PBMC: https://satijalab.org/azimuth/[2] 单细胞转录组数据分析||Seurat新版教程: Integration and Label Transfer: https://www.jianshu.com/p/d43f16bdfed9[3] https://satijalab.org/seurat/v4.0/reference_mapping.html
 往期回顾
往期回顾
RNA-seq的3的差异分析R包你选择哪个
原汁原味读 | 单细胞肿瘤免疫图谱
CNS图表复现17—inferCNV结果解读及利用之进阶
如果你对单细胞转录组研究感兴趣,但又不知道如何入门,也许你可以关注一下下面的课程
单细胞初级8讲和高级分析8讲
老板,请为我配备一个懂生信的师兄
你以为GEO只是挖挖就完了吗


看完记得顺手点个“在看”哦!
 生物 | 单细胞 | 转录组丨资料
每天都精彩
生物 | 单细胞 | 转录组丨资料
每天都精彩
长按扫码可关注
最后
以上就是英勇雪碧最近收集整理的关于西瓜数据集4.0_Seurat 4.0 || 单细胞PBMC多模态参考数据集前情回顾正文的全部内容,更多相关西瓜数据集4.0_Seurat内容请搜索靠谱客的其他文章。







发表评论 取消回复