此文章是通过学习瑞典国家生物信息学基础设施(NBIS) 所开放的单细胞分析教程加上网上所查找的资料,自身的理解所形成的,可能会有不足之处。该部分是使用整合后的PCA来进行聚类。
参考来源:https://nbisweden.github.io/workshop-scRNAseq/labs/compiled/seurat/seurat_01_qc.html
感兴趣的话可以阅读原文。
聚类分析
首先构建neighbour graph ,以便后续在图上进行聚类,这里将展示层次聚类与k-means聚类。
if (!require(clustree)) {
install.packages("clustree", dependencies = FALSE)
}
suppressPackageStartupMessages({
library(Seurat)
library(cowplot)
library(ggplot2)
library(pheatmap)
library(rafalib)
library(clustree)
})
alldata <- readRDS("data/results/covid_qc_dr_int.rds")
构图聚类
在图上的聚类一共分为三步:
- 根据数据构建KNN图。
- 修剪KNN中的虚拟链接,使其成为一个SNN图。
- 找到与其他组之间的链接相比,组内链接最大化的单元组。
构建KNN
# 检查现在的默认数据集是否还是CCA
alldata@active.assay
# [1] "CCA"
alldata <- FindNeighbors(alldata, dims = 1:30, k.param = 60, prune.SNN = 1/15)
names(alldata@graphs)
# [1] "CCA_nn" "CCA_snn"
# 查看连接之间的大小
alldata@graphs$CCA_nn[1:10,1:10]
# 10 x 10 sparse Matrix of class "dgCMatrix"
# [[ suppressing 10 column names ‘covid_1_AGGGTCCCATGACCCG-1’, ‘covid_1_TACCCACAGCGGGTTA-1’, ‘covid_1_CCCAACTTCATATGGC-1’ ... ]]
#
# covid_1_AGGGTCCCATGACCCG-1 1 . . . . . . . . 1
# covid_1_TACCCACAGCGGGTTA-1 . 1 . . . . . . . .
# covid_1_CCCAACTTCATATGGC-1 . . 1 . . . . . . .
# covid_1_TCAAGTGTCCGAACGC-1 . . . 1 1 . . . . .
# covid_1_ATTCCTAGTGACTGTT-1 . . . . 1 . . . . .
# covid_1_GTGTTCCGTGGGCTCT-1 . . . . . 1 . . . .
# covid_1_CCTAAGACAGATTAAG-1 . . . . . . 1 . . .
# covid_1_AATAGAGAGGGTTAGC-1 . . . . . . . 1 1 .
# covid_1_GGGTCACTCACCTACC-1 . . . . . . . 1 1 .
# covid_1_TCCTCTTGTACAGTCT-1 1 . . . . . . . . 1
# 再绘制热查看
pheatmap(alldata@graphs$CCA_nn[1:200, 1:200], col = c("white", "black"), border_color = "grey90",
legend = F, cluster_rows = F, cluster_cols = F, fontsize = 2)

从上面可以看得出来,其中每个单元之间的连接都表示为1,是一个定值,这被称为未加权图(Seurat系统默认)。然而,在实际情况下,单元之间的连接重要程度是不一样的,一些单元之间的内部连接可能会比这些单元与其他单元的连接更加重要,在这种情况下,图表的比例可以从0开始直到最大距离。通常情况下,两点之间距离越小,代表他们之间的联系越紧密,这种图被称为加权图。加权图与未加权图都适合聚类,但是对于大型数据集(> 100k 单元),未加权图上聚类更快。
图上聚类
构建图完成之后,就可以执行图聚类。聚类是根据你指定的分辨率完成的,分辨率可以解释为你想要集群能够代表的细胞精细程度,就是你想要分到多具体的细胞,分辨率越高,所分得到的细胞类型越多。
在Seurat中,函数FindClusters默认使用Louvain算法(algorithm=1),进行图上进行聚类,而如果需要使用leiden算法(algorithm=4)。
# 使用Louvain进行不同分辨率的聚类
for (res in c(0.1, 0.25, 0.5, 1, 1.5, 2)) {
alldata <- FindClusters(alldata, graph.name = "CCA_snn", resolution = res, algorithm = 1)
}
# 每次运行聚类时,数据都储存在元数据中
plot_grid(ncol = 3, DimPlot(alldata, reduction = "umap", group.by = "CCA_snn_res.0.5") +
ggtitle("louvain_0.5"), DimPlot(alldata, reduction = "umap", group.by = "CCA_snn_res.1") +
ggtitle("louvain_1"), DimPlot(alldata, reduction = "umap", group.by = "CCA_snn_res.2") +
ggtitle("louvain_2"))
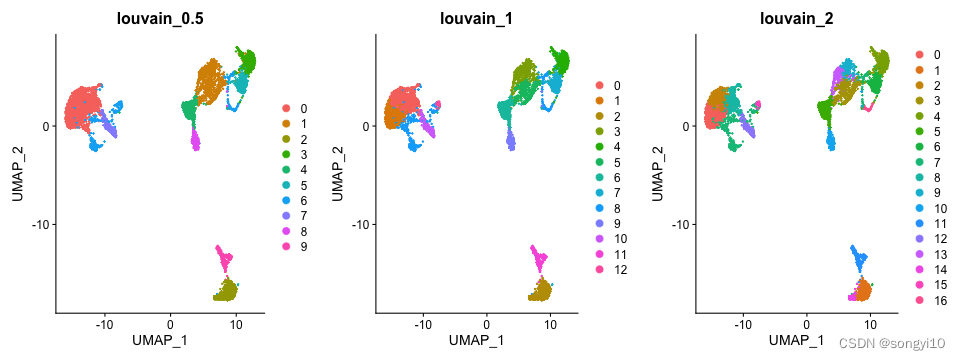
我们现在可以使用 clustree 包来可视化细胞如何根据分辨率在clustering之间分布。
# install.packages('clustree')
suppressPackageStartupMessages(library(clustree))
clustree(alldata@meta.data, prefix = "CCA_snn_res.")
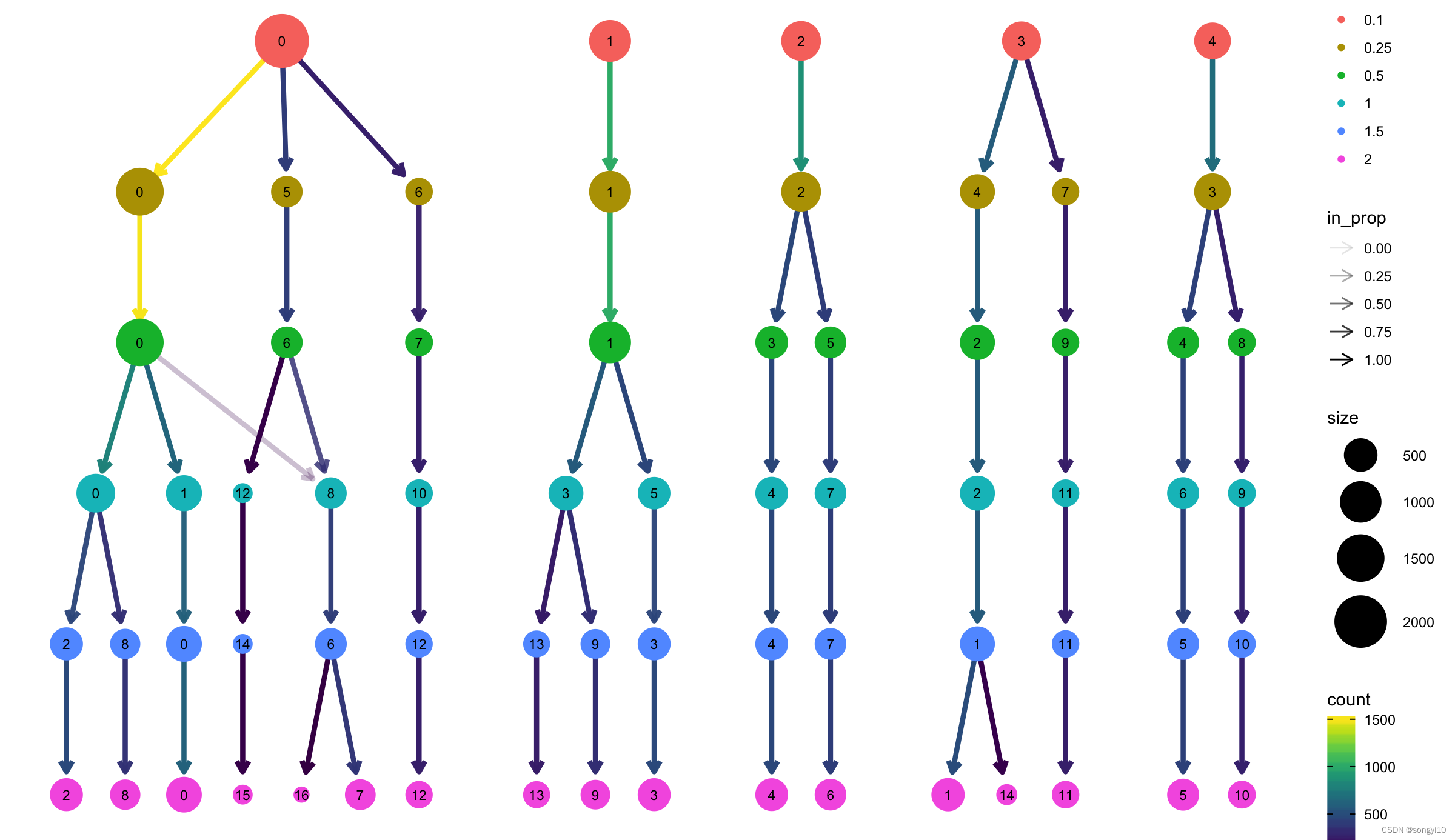
可以从上面这几张图看出来分辨率与集群(clustering)数量之间的关系。
k-means 聚类
K-means 是一种通用的聚类算法,通常,它应用于表达式数据的降维表示(最常见的是 PCA,因为低维距离的可解释性)。我们需要提前定义集群 k 的数量。由于结果取决于集群中心的初始化(k的设定),因此通常建议运行具有多个启动配置的 K-means(通过 nstart 参数)。
for (k in c(5, 7, 10, 12, 15, 17, 20)) {
alldata@meta.data[, paste0("kmeans_", k)] <- kmeans(x = alldata@reductions[["pca"]]@cell.embeddings,centers = k, nstart = 100)$cluster
}
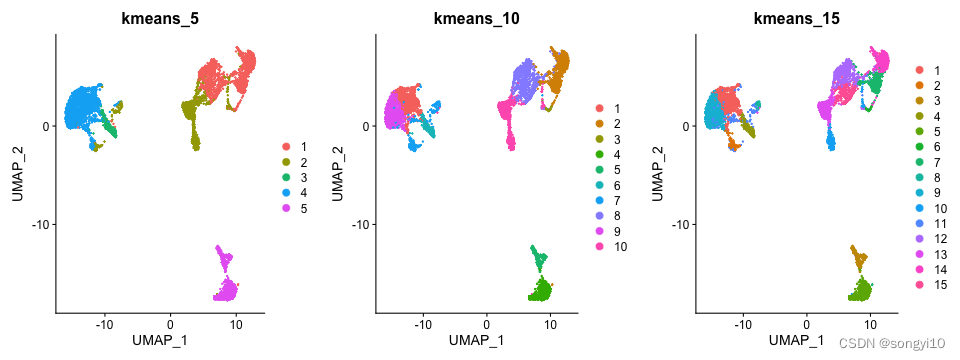
plot_grid(ncol = 3, DimPlot(alldata, reduction = "umap", group.by = "kmeans_5") +
ggtitle("kmeans_5"), DimPlot(alldata, reduction = "umap", group.by = "kmeans_10") +
ggtitle("kmeans_10"), DimPlot(alldata, reduction = "umap", group.by = "kmeans_15") +
ggtitle("kmeans_15"))
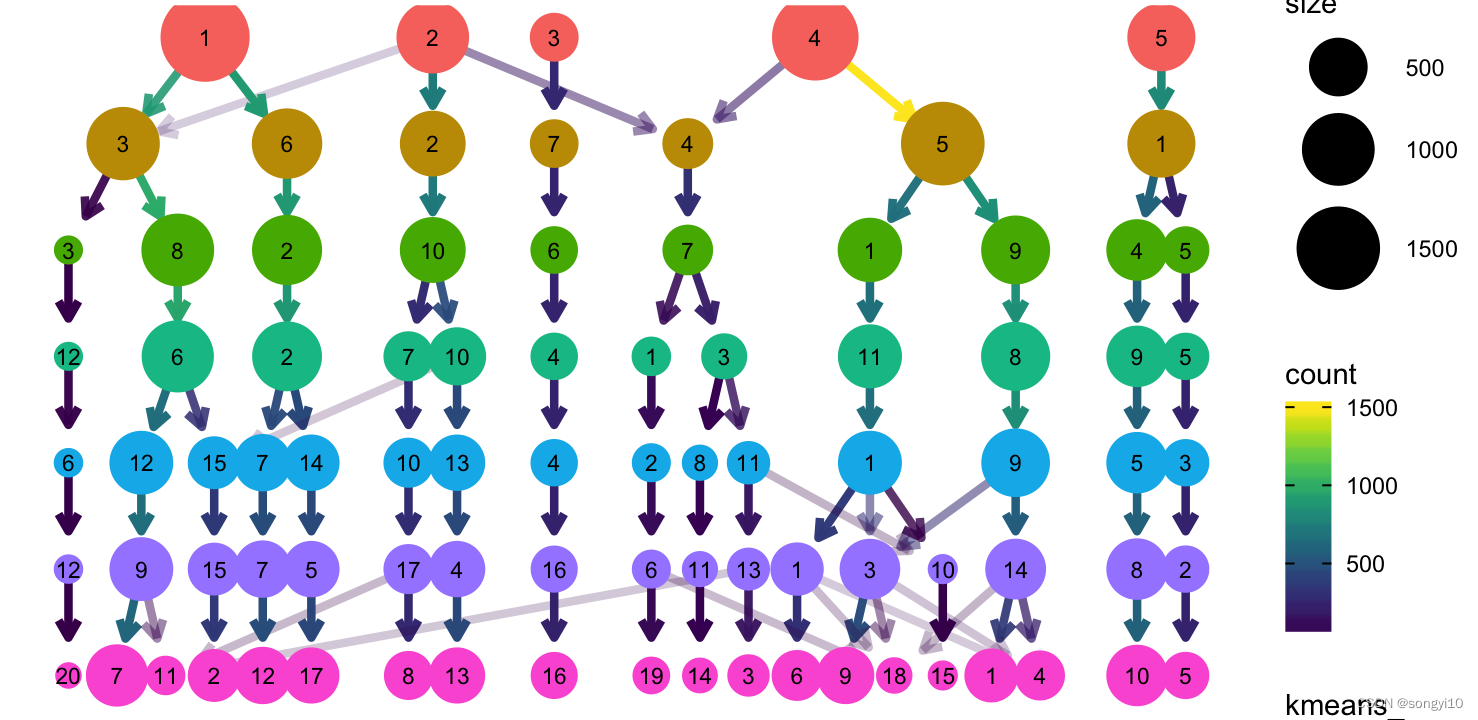
clustree(alldata@meta.data, prefix = "kmeans_")
层次聚类
距离矩阵
在R基础包stat中已经包含了一个可以计算所有样本对之间的距离的函数dist(),dist()函数可用的距离有,“euclidean”、“maximum”、“manhattan”、“canberra”、“binary”或“minkowski”等。。因为我们需要的是细胞之间的距离,所以,需要对原始的表达矩阵进行转置。
d <- dist(t(alldata@reductions[["pca"]]@cell.embeddings), method = "euclidean")
除了dist()计算的距离以外,我们还可以利用cor()计算细胞之间的相关性从而评估细胞之间的联系,但是相关性系数是从-1到1,其中小于0的数值在距离上并没有意义,所以,这个时候,我们需要做一些转换,把相关性系数转换为距离对象,这时候我们通过一个公式
a
d
j
=
1
−
c
o
r
2
adj = frac{1- cor}{2}
adj=21−cor ,将其转换为邻接矩阵。
通过这样的 转换,我们就可以使用as.dist()函数将这个比例转换为距离对象,最大值为1,即最不相关,最小值为0,即最相关。
# 计算相关性
sample_cor <- cor(Matrix::t(alldata@reductions[["pca"]]@cell.embeddings))
# 转换为邻接矩阵
sample_cor <- (1 - sample_cor)/2
# 转换为距离对象
d2 <- as.dist(sample_cor)
层次聚类
在计算完距离之后,就可以使用hclust函数进行层次聚类了,其中可以使用的方法有:“ward.D”、“ward.D2”、“single”、“complete”、“average”、“mcquitty”、“median”或“centroid”。这里仅展示"ward.D2"方法。
# euclidean,欧氏距离
h_euclidean <- hclust(d, method = "ward.D2")
# correlation,相关性距离
h_correlation <- hclust(d2, method = "ward.D2")
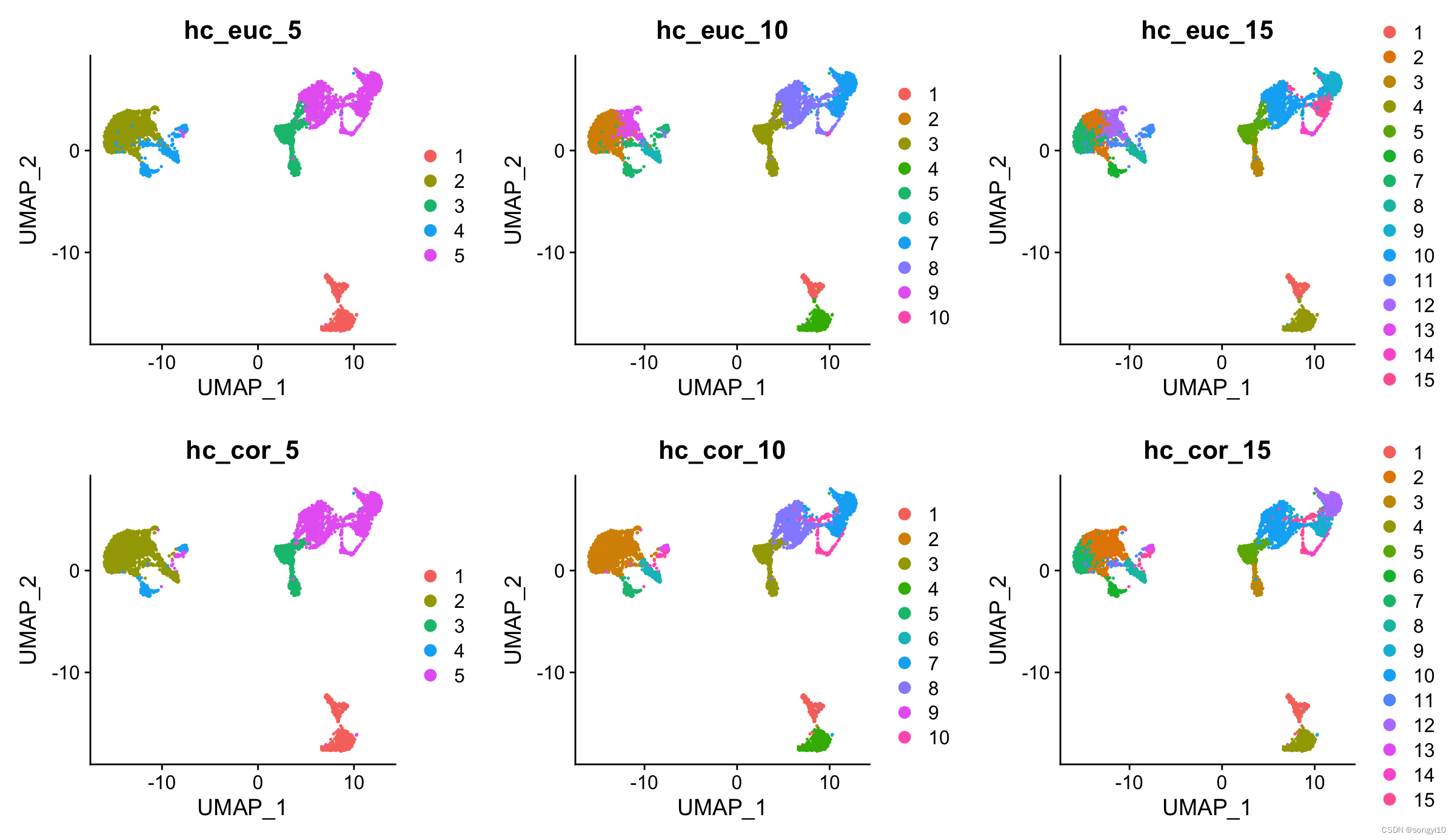
创建完树状图后,我们紧接着就是去定义哪些样本属于哪些集群,即在不同的级别以固定的阈值(cuttree)切割树以定义集群,从而定义集群的数量和高度,也可以尝试不同的集群。
#euclidean distance
alldata$hc_euclidean_5 <- cutree(h_euclidean,k = 5)
alldata$hc_euclidean_10 <- cutree(h_euclidean,k = 10)
alldata$hc_euclidean_15 <- cutree(h_euclidean,k = 15)
#correlation distance
alldata$hc_corelation_5 <- cutree(h_correlation,k = 5)
alldata$hc_corelation_10 <- cutree(h_correlation,k = 10)
alldata$hc_corelation_15 <- cutree(h_correlation,k = 15)
plot_grid(ncol = 3,
DimPlot(alldata, reduction = "umap", group.by = "hc_euclidean_5")+ggtitle("hc_euc_5"),
DimPlot(alldata, reduction = "umap", group.by = "hc_euclidean_10")+ggtitle("hc_euc_10"),
DimPlot(alldata, reduction = "umap", group.by = "hc_euclidean_15")+ggtitle("hc_euc_15"),
DimPlot(alldata, reduction = "umap", group.by = "hc_corelation_5")+ggtitle("hc_cor_5"),
DimPlot(alldata, reduction = "umap", group.by = "hc_corelation_10")+ggtitle("hc_cor_10"),
DimPlot(alldata, reduction = "umap", group.by = "hc_corelation_15")+ggtitle("hc_cor_15")
保存数据
saveRDS(alldata, "data/results/covid_qc_dr_int_cl.rds")
最后
以上就是深情雨最近收集整理的关于NBIS单细胞教程:降维聚类(四)的全部内容,更多相关NBIS单细胞教程内容请搜索靠谱客的其他文章。








发表评论 取消回复