继续搬一点近期飞书文档模拟的到博客里
参考博客:
- Gromac中文教程:https://jerkwin.github.io/GMX/GMXtut-5/#%E6%A6%82%E8%BF%B0
- https://www.jianshu.com/p/b10fe4b4af11
- https://www.bilibili.com/read/cv6496041/
生成小分子过程
- 生成:charm gui 官网:https://charmm-gui.org/?doc=input/pdbreader&step=0
- PDBID:6kyk,6kyh。生成时注意选择第一个。
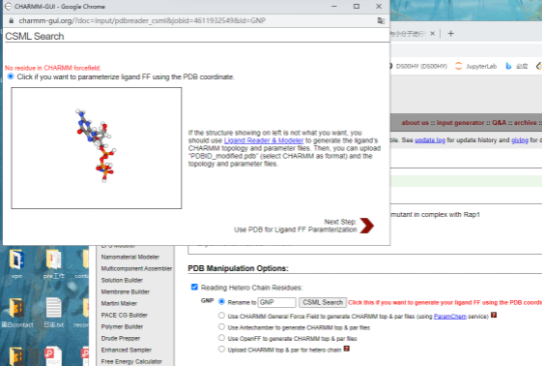
- 在GNP文件中的mol2
- CGenFF上传 mol2:https://cgenff.umaryland.edu/userAccount/userLogin.php#20220227_5/6kyk_gnp.err。
- 下载str,生成prm、itp、pdb
python cgenff_charmm2gmx_py3_nx2.py GNP GNP.mol2 gnp.str charmm36-mar2019.ff- 注意NetworkX版本,若是最新版不一致,将self.G.node替换成新版语法self.G.nodes
- 力场下载地址:http://mackerell.umaryland.edu/charmm_ff.shtml#charmm
- 生成gro文件
gmx pdb2gmx -ignh -ff charmm36-mar2019 -f 6kyh_mg.pdb -o 6kyh.gro -p 6kyh.top -water tip3p
gmx editconf -f gnp_ini.pdb -o gnp.gro
- 将小分子gro坐标添加进6kyh.gro,修改gro原子数,32399 prm,itp文件include到6kyh.top,,增加GNP 1到top。一定要注意include prm文件的位置,一定要在所有的分子itp文件之前,力场文件之下,否则会导致GROMACS无法识别加入的小分子。
# add gro , +49
# vim 6kyh.top,注意位置,prm一定要在所有的itp之前,而在力场之下。
; Include ligand topology
#include "gnp.prm"
#include "gnp.itp"模拟
-
水盒子
gmx editconf -f 6kyh.gro -o 6kyh-PBC.gro -bt dodecahedron -d 1.0
-
真空最小化
gmx grompp -f em-vac-pme.mdp -c 6kyh-PBC.gro -p 6kyh.top -o em-vac.tpr
gmx mdrun -v -deffnm em-vac
- em-vac-pme.mdp
; 传递给预处理器的一些定义
define = -DFLEXIBLE ; 使用柔性水模型而非刚性模型, 这样最陡下降法可进一步最小化能量
; 模拟类型, 结束控制, 输出控制参数
integrator = steep ; 指定使用最陡下降法进行能量最小化. 若设为`cg`则使用共轭梯度法
emtol = 500.0 ; 若力的最大值小于此值则认为能量最小化收敛(单位kJ mol^-1^ nm^-1^)
emstep = 0.01 ; 初始步长(nm)
nsteps = 50000 ; 在能量最小化中, 指定最大迭代次数
nstenergy = 1 ; 能量写出频率
energygrps = System ; 要写出的能量组
; 近邻列表, 相互作用计算参数
nstlist = 1 ; 更新近邻列表的频率. 1表示每步都更新
ns_type = grid ; 近邻列表确定方法(simple或grid)
; ;coulombtype = PME ; ;无离子会报错 计算长程静电的方法. PME为粒子网格Ewald方法, 还可以使用cut-off
cutoff-scheme = Verlet
rlist = 1.0 ; 短程力近邻列表的截断值
rcoulomb = 1.0 ; 长程库仑力的截断值
vdwtype = cut-off ; 计算范德华作用的方法
rvdw = 1.0 ; 范德华距离截断值
constraints = none ; 设置模型中使用的约束
pbc = xyz ; 3维周期性边界条件-
溶剂和离子
# 溶剂和离子 选择SOL,echo19
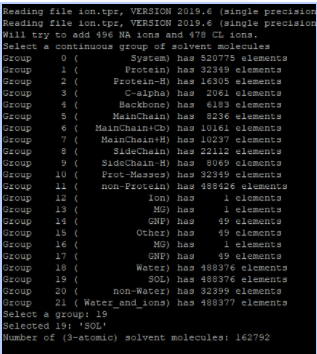
gmx solvate -cp em-vac.gro -cs spc216.gro -p 6kyh.top -o 6kyh-b4ion.gro
gmx grompp -f em-sol-pme.mdp -c 6kyh-b4ion.gro -p 6kyh.top -o ion.tpr
echo 19 | gmx genion -s ion.tpr -o 6kyh-b4em.gro -neutral -conc 0.15 -p 6kyh.top
注意-conc选项的离子浓度,需要根据实验设定进行设计,可以选择默认浓度。
- em-sol-pme.mdp
define = -DFLEXIBLE
integrator = steep
emtol = 250.0
nsteps = 50000
nstenergy = 1
energygrps = System
nstlist = 1
ns_type = grid
;; coulombtype = PME ;无离子会报错
rlist = 1.0
rcoulomb = 1.0
rvdw = 1.0
constraints = none
pbc = xyz-
能量最小化
-
再次生成tpr文件,并运行能量最小化
#能量最小化
gmx grompp -f em-sol-pme.mdp -c 6kyh-b4em.gro -p 6kyh.top -o em-sol.tpr
gmx mdrun -v -deffnm em-sol
-
限制+NVT平衡
- 是否需要限制配体,(此步根据需求来,对于我的实验可以略过。)
gmx make_ndx -f em.gro -o index.ndx
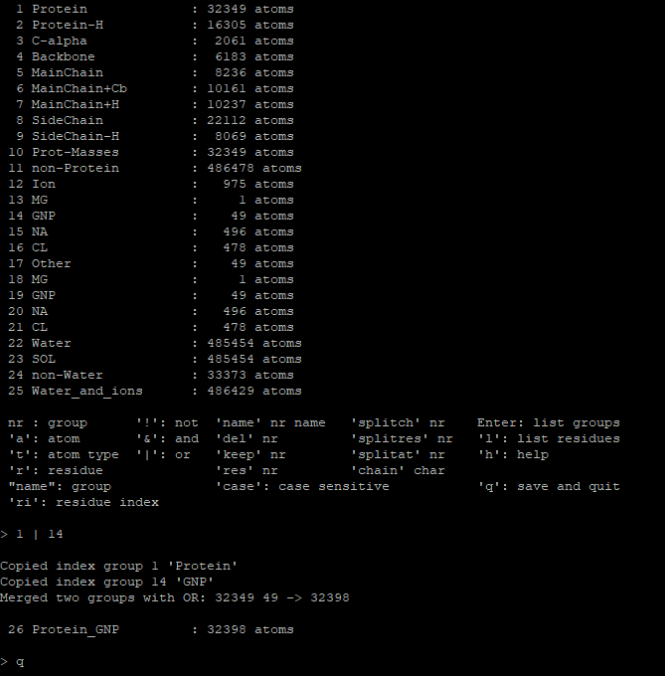
-
设置
tc_grps = Protein_JZ4 Water_and_ions来达到我们需要的Protein Non-Protein
的效果了,模拟时要加-n index.ndx
- 平衡
#NVT平衡
##设置 温度耦合分组,
gmx grompp -f nvt-pr-md.mdp -r em-sol.gro -c em-sol.gro -p 6kyh.top -o nvt-pr.tpr
gmx mdrun -deffnm nvt-pr
- nvt-pr-md.mdp
预处理选项
define = -DPOSRES ; 告诉GROMACS运行位置限制性模拟
; 运行控制参数
integrator = md
dt = 0.002 ; 时间步长(单位为ps, 我们使用了2 fs). 只用于动力学积分器(如md), 能量最小化时不需要
nsteps = 5000000 ; ; 10ns模拟步数(总模拟时间为nsteps*dt)
; 输出控制参数
nstxout = 50000 ;; 要100ps 输出模拟坐标的频率(nstxout=500且dt=0.002, 所以每1 ps输出一次)
nstvout = 50000 ; 速度保存频率
nstenergy = 50000 ; 能量保存频率
nstlog = 50000 ; log文件输出频率
energygrps = Protein Non-Protein
; 近邻列表参数
nstlist = 5
ns_type = grid
pbc = xyz
rlist = 1.0
; 静电和VDW参数
coulombtype = PME ; 长程静电相互作用的计算方法
pme_order = 4 ; 三次插值
fourierspacing = 0.16 ; FFT间隔
rcoulomb = 1.0 ; 计算静电作用的截断值(单位nm)
vdw-type = Cut-off
rvdw = 1.0
; 温度耦合部分非常重要, 必须正确填写.
tcoupl = v-rescale ; 随机重新调整速度
tc-grps = Protein Non-Protein ; 与控温器耦合的组(模型中的每个原子或残基都用一定的索引组表示), 对蛋白和非蛋白使用不同的组分开控制
tau_t = 0.1 0.1 ; 温度耦合的时间常数(单位ps). 必须每个tc_grps指定一个, 且顺序对应
ref_t = 300 300 ; 代表耦合的参考温度(即动力学模拟的温度, 单位K). 每个tc_grp对应一个ref_t
; 色散校正
DispCorr = EnerPres ; 校正VDW截断
; 不使用压力耦合
pcoupl = no ; NVT中不能使用压力耦合
; 初始速度选项
gen_vel = yes ; 根据Maxwell分布随机产生速度
gen_temp = 300 ; 当你改变温度时, 别忘了改变gen_temp变量以生成速度
gen_seed = -1 ; 随机数生成器的种子
; 键约束选项
constraints = all-bonds ; 使用LINCS算法约束所有键
continuation = no ; 第一次运行
constraint_algorithm = lincs ; 约束算法
lincs_iter = 1 ; LINCS精度
lincs_order = 4 ; LINCS阶数, 与精度有关-
NPT平衡
#
gmx grompp -f npt-pr-md.mdp -r nvt-pr.gro -c nvt-pr.gro -p 6kyh.top -o npt-pr.tpr
gmx mdrun -deffnm npt-pr
- npt-pr-md.mdp
define = -DPOSRES
integrator = md
dt = 0.002
nsteps = 5000000 ;10ns
nstxout = 50000 ;100ps
nstvout = 50000
nstfout = 50000
nstenergy = 50000
nstlog = 50000
energygrps = Protein Non-Protein
nstlist = 5
ns-type = Grid
pbc = xyz
rlist = 1.0
coulombtype = PME
pme_order = 4
fourierspacing = 0.16
rcoulomb = 1.0
vdw-type = Cut-off
rvdw = 1.0
Tcoupl = v-rescale
tc-grps = Protein Non-Protein
tau_t = 0.1 0.1
ref_t = 300 300
DispCorr = EnerPres
; 压力耦合
Pcoupl = Parrinello-Rahman ; Parrinello-Rahman控压器.
Pcoupltype = Isotropic ; isotropic 指盒子可以平均地向各个方向(x, y,z)膨胀或压缩以维持一定的压力. 进行膜模拟时需要用semiisotropic.
tau_p = 2.0 ; 压力耦合的时间常数(单位ps).
compressibility = 4.5e-5 ; 溶剂的压缩系数(4.5e-5为水在300 K和标准大气压下的压缩系数).
ref_p = 1.0 ; 压力耦合的参考压力(单位bar, 1大气压约为0.983 bar).
refcoord_scaling = com
gen_vel = no ; 不产生速度
constraints = all-bonds
continuation = yes
constraint_algorithm = lincs
lincs_iter = 1
lincs_order = 4-
成品模拟
gmx grompp -f npt-nopr-md.mdp -c npt-pr.gro -p 6kyh.top -o npt-nopr.tpr
gmx mdrun -deffnm npt-nopr
- npt-nopr-md.mdp
integrator = md
dt = 0.002
nsteps = 500000 ; ;cd
nstxout = 50000 ;100ps
nstvout = 50000
nstfout = 50000
nstenergy = 50000
nstlog = 500 ; log要小10psls
energygrps = Protein Non-Protein
nstlist = 10
ns-type = Grid
pbc = xyz
rlist = 1.0
coulombtype = PME
pme_order = 4
fourierspacing = 0.16
rcoulomb = 1.0
vdw-type = Cut-off
rvdw = 1.0
Tcoupl = v-rescale
tc-grps = Protein Non-Protein
tau_t = 0.1 0.1
ref_t = 300 300
DispCorr = EnerPres
Pcoupl = Parrinello-Rahman
Pcoupltype = Isotropic
tau_p = 2.0
compressibility = 4.5e-5
ref_p = 1.0
gen_vel = no
constraints = all-bonds
continuation = yes
constraint_algorithm = lincs
lincs_iter = 1
lincs_order = 4报错
- No,
- 修改:
; Include ligand topology
#include "gnp.top"
- 网上建议:https://www.researchgate.net/post/How_to_solve_the_no_default_type_errors_in_GROMACS-507_using_CHARMM36ff
- 修改一下力场版本:
- 最终解决方案:http://www.gromacs.org/Documentation/Errors#Invalid_order_for_directive_xxx
6kyk
python cgenff_charmm2gmx_py3_nx2.py GNP GNP.mol2 gnp.str charmm36-mar2019.ff
gmx pdb2gmx -ignh -ff charmm36-mar2019 -f 6kyk_mg.pdb -o 6kyk.gro -p 6kyk.top -water tip3p
gmx editconf -f gnp_ini.pdb -o gnp.gro
; 21844+49=21893
;Include ligand topology
#include "gnp.prm"
#include "gnp.itp"
gmx editconf -f 6kyk.gro -o 6kyk-PBC.gro -bt dodecahedron -d 1.0
gmx grompp -f em-vac-pme.mdp -c 6kyk-PBC.gro -p 6kyk.top -o em-vac.tpr
gmx mdrun -v -deffnm em-vac
gmx solvate -cp em-vac.gro -cs spc216.gro -p 6kyk.top -o 6kyk-b4ion.gro
gmx grompp -f em-sol-pme.mdp -c 6kyk-b4ion.gro -p 6kyk.top -o ion.tpr
echo 19 | gmx genion -s ion.tpr -o 6kyk-b4em.gro -neutral -conc 0.15 -p 6kyk.top
gmx grompp -f em-sol-pme.mdp -c 6kyk-b4em.gro -p 6kyk.top -o em-sol.tpr
gmx mdrun -v -deffnm em-sol
gmx grompp -f nvt-pr-md.mdp -r em-sol.gro -c em-sol.gro -p 6kyk.top -o nvt-pr.tpr
gmx mdrun -deffnm nvt-pr
gmx grompp -f npt-pr-md.mdp -r nvt-pr.gro -c nvt-pr.gro -p 6kyk.top -o npt-pr.tpr
gmx mdrun -deffnm npt-pr
gmx grompp -f npt-nopr-md.mdp -c npt-pr.gro -p 6kyk.top -o npt-nopr.tpr
gmx mdrun -deffnm npt-nopr速度测试
- 深圳超算(速度会有0.x ns的波动)
| PDB | 总核数 | 速度ns/day | 模拟时间 | 盒子大小 |
| 6kyh | 60 | 4.66 | 400ps | 1.0 |
| 96 | 5.452 | |||
| 120 | 5.783 | |||
| 144 | 5.693 | |||
| 180 | 6.7 | |||
| 6kyk | 96 | 9.8 | ||
| 120 | 10.9 | |||
| 144 | 11.3 |
| PDB | 总核数 | 速度ns/day | 盒子大小 |
| 6kyh | 96 | 5.749 | 0.8 |
| 5.452 | 1.0 | ||
| 5.494 | 1.2 | ||
| 6kyk | 96 | 10.241 | 0.8 |
| 9.881 | 1.0 | ||
| 9.776 | 1.2 |
存储大小
- 400ps,100ps存一次,分别为90M和48M。
最后
以上就是留胡子高跟鞋最近收集整理的关于蛋白+小分子配体md(详细保姆教程)参考博客:生成小分子过程模拟报错6kyk速度测试存储大小的全部内容,更多相关蛋白+小分子配体md(详细保姆教程)参考博客内容请搜索靠谱客的其他文章。








发表评论 取消回复