之前我们完成了 接着就是最重要的环节了,将不同 不得不说,东西还是挺多的,而且非常重要,我们一起来试一下吧。???? 这里我们需要将 这里我们需要对模块的 用相关性矩阵可视化一下吧。???? 1️⃣ 接着我们将 2️⃣ 这里假设我们感兴趣的是 对于我们找到的有意义的模块,可以进一步的分析模块内部的基因,具体是哪个基因在其中更为重要。???? 当然,这就要用到我们之前计算好的 这里我们假设感兴趣的是 可能你也直接输出所有模块的结果,然后再挑选你需要的,那就用这段批量输出的代码吧。???? ???? 点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰 ???? ???? ComplexHeatmap | 颜狗写的高颜值热图代码!1写在前面
WGCNA输入数据的清洗,网络构建和模块识别。????
而且还介绍了如何对大型数据分级处理,有效地减少了内存的负担。????
module与表型或者临床特征相联系,进一步鉴定出有意义的module,并进行module内部的分析,筛选重要基因。????2用到的包
rm(list = ls())
library(WGCNA)
library(tidyverse)3示例数据
load("FemaleLiver-01-dataInput.RData")
load("FemaleLiver-02-networkConstruction-auto.RData")4模块与外部特征关联
module和traits联系起来,并且采用量化的方式。????4.1 量化模块与特征之间的关系
eigengenes进行提取,并与traits进行相关性分析。????nGenes <- ncol(datExpr)
nSamples <- nrow(datExpr)
MEs0 <- moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs <- orderMEs(MEs0)
moduleTraitCor <- cor(MEs, datTraits, use = "p")
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nSamples)
sizeGrWindow(10,6)
textMatrix = paste(signif(moduleTraitCor, 2), "n(",
signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(6, 8.5, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))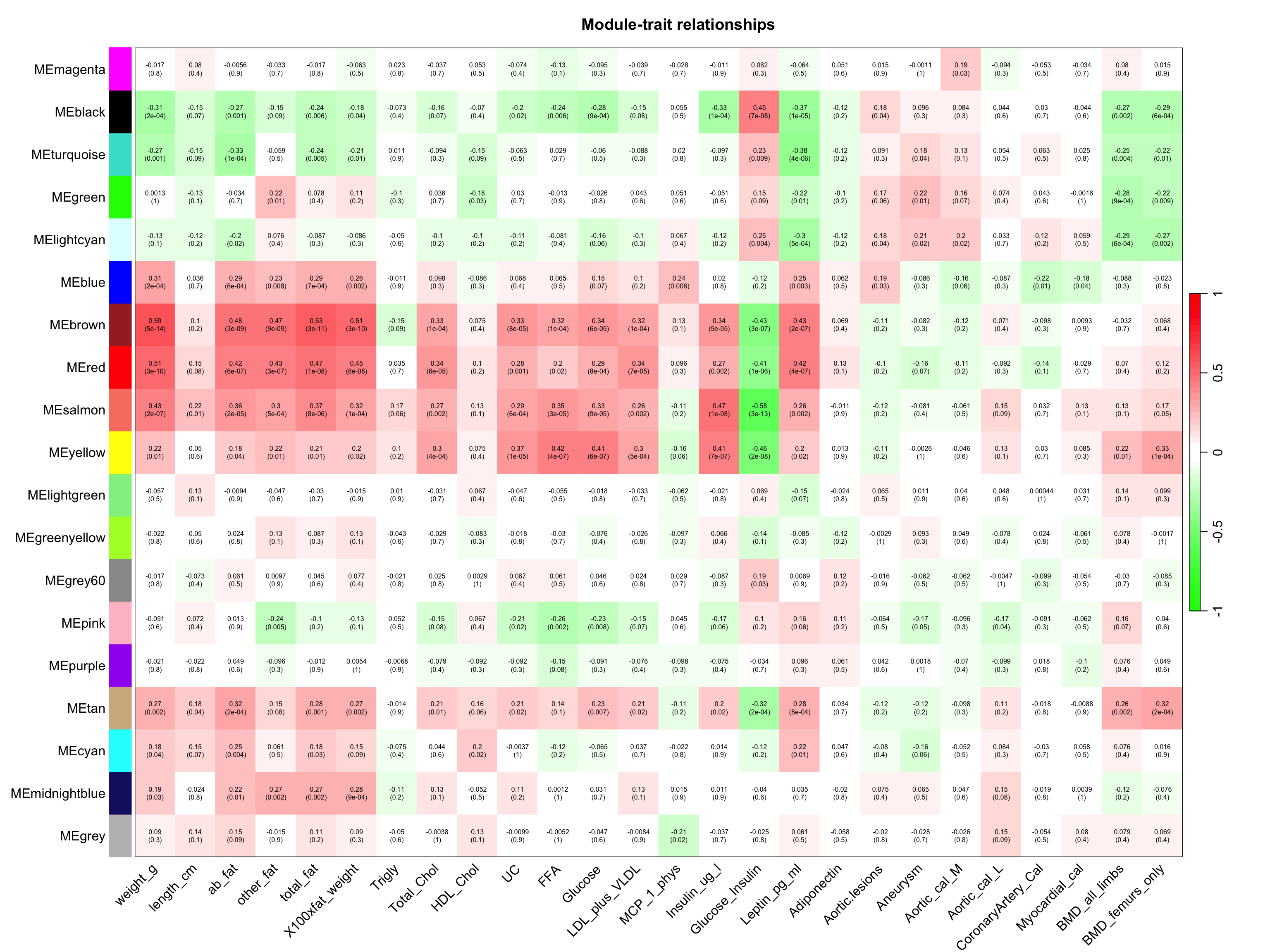
4.2 计算Gene Significance 和 Module Membership
Gene Significance(GS) 定义为量化基因与traits之间相关性的绝对值。
Module Membership(MM)定义为模块的eigengene与基因表达谱之间的相关性。
weight这个特征,想找到与weight相关的module以及其中的基因。????weight <- as.data.frame(datTraits$weight_g);
names(weight) <- "weight"
modNames <- substring(names(MEs), 3)
geneModuleMembership <- as.data.frame(cor(datExpr, MEs, use = "p"))
MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples))
names(geneModuleMembership) <- paste("MM", modNames, sep="")
names(MMPvalue) <- paste("p.MM", modNames, sep="")
geneTraitSignificance <- as.data.frame(cor(datExpr, weight, use = "p"))
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
names(geneTraitSignificance) <- paste("GS.", names(weight), sep="")
names(GSPvalue) <- paste("p.GS.", names(weight), sep="")
4.3 模块内部分析
GS和MM了。????magenta这个模块吧。????module <- "magenta"
column <- match(module, modNames)
moduleGenes <- moduleColors==module
sizeGrWindow(7, 7)
par(mfrow = c(1,1))
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = "Gene significance for body weight",
main = paste("Module membership vs. gene significancen"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)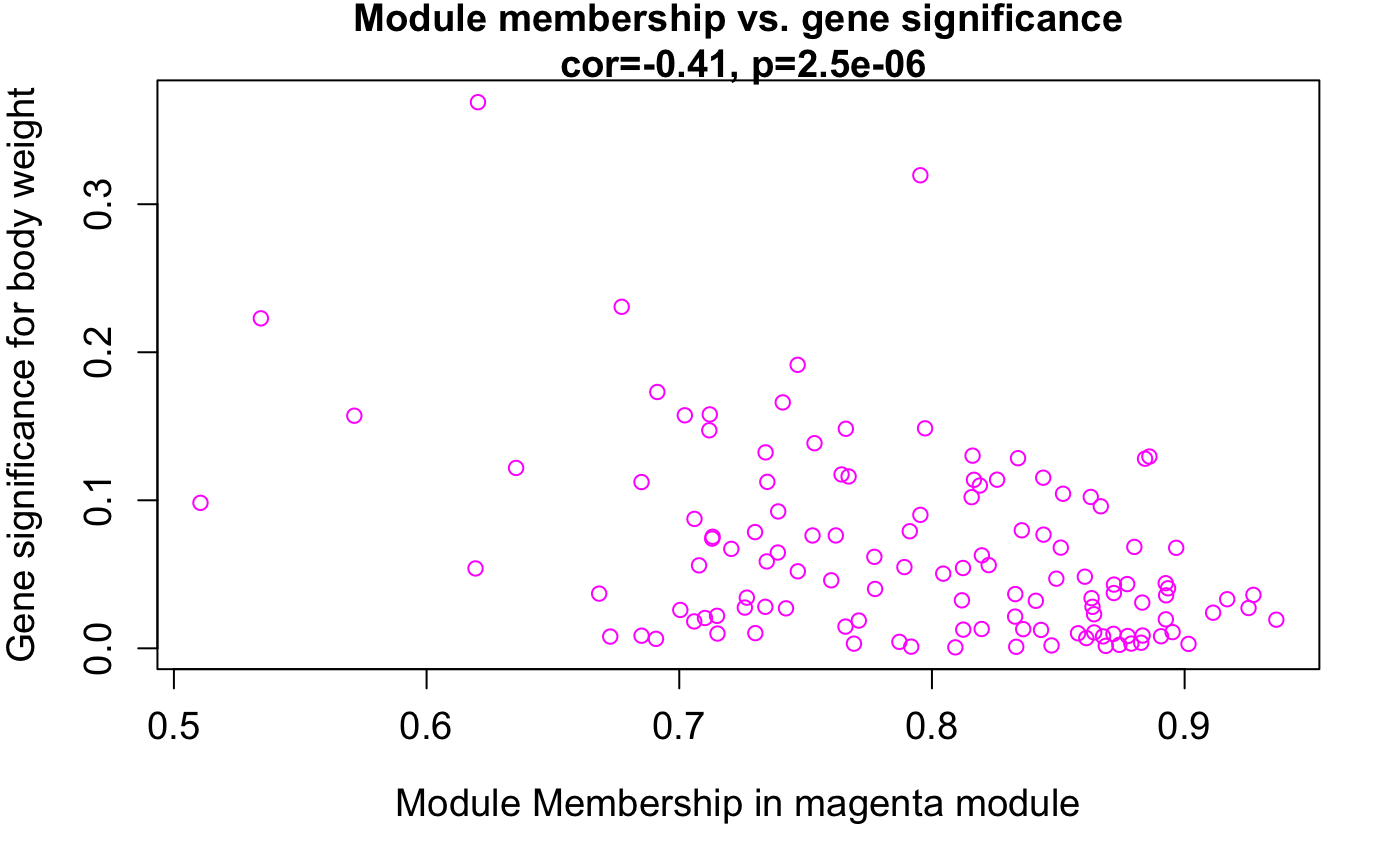
4.4 批量输出
modNames <- substring(names(MEs), 3)
geneModuleMembership <- as.data.frame(cor(datExpr, MEs, use = "p"))
MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples))
names(geneModuleMembership) <- paste("MM", modNames, sep="")
names(MMPvalue) = paste("p.MM", modNames, sep="")
traitNames <- names(datTraits)
geneTraitSignificance <- as.data.frame(cor(datExpr, datTraits, use = "p"))
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
names(geneTraitSignificance) <- paste("GS.", traitNames, sep="")
names(GSPvalue) <- paste("p.GS.", traitNames, sep="")
for (trait in traitNames){
traitColumn = match(trait,traitNames)
for (module2 in modNames){
column = match(module2, modNames)
moduleGenes = moduleColors==module2
if (nrow(geneModuleMembership[moduleGenes,]) > 1){
pdf(file = paste0("./module_", trait, "_", module,".pdf"),
width=7,height=7)
par(mfrow = c(1,1))
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, traitColumn]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for ",trait),
main = paste("Module membership vs. gene significancen"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
dev.off()
}
}
}
5结果汇总输出
5.1 读入并整理注释文件
annot <- read.csv(file = "./FemaleLiver-Data/GeneAnnotation.csv");
dim(annot)
names(annot)
probes <- names(datExpr)
probes2annot <- match(probes, annot$substanceBXH)
sum(is.na(probes2annot))
5.2 整理并输出结果文件
geneInfo0 <- data.frame(substanceBXH = probes,
geneSymbol = annot$gene_symbol[probes2annot],
LocusLinkID = annot$LocusLinkID[probes2annot],
moduleColor = moduleColors,
geneTraitSignificance,
GSPvalue)
modOrder <- order(-abs(cor(MEs, weight, use = "p")))
for (mod in 1:ncol(geneModuleMembership))
{
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneModuleMembership[, modOrder[mod]],
MMPvalue[, modOrder[mod]]);
names(geneInfo0) = c(oldNames, paste("MM.", modNames[modOrder[mod]], sep=""),
paste("p.MM.", modNames[modOrder[mod]], sep=""))
}
geneOrder <- order(geneInfo0$moduleColor, -abs(geneInfo0$GS.weight));
geneInfo <- geneInfo0[geneOrder, ]
write.csv(geneInfo, file = "geneInfo.csv")
DT::datatable(geneInfo)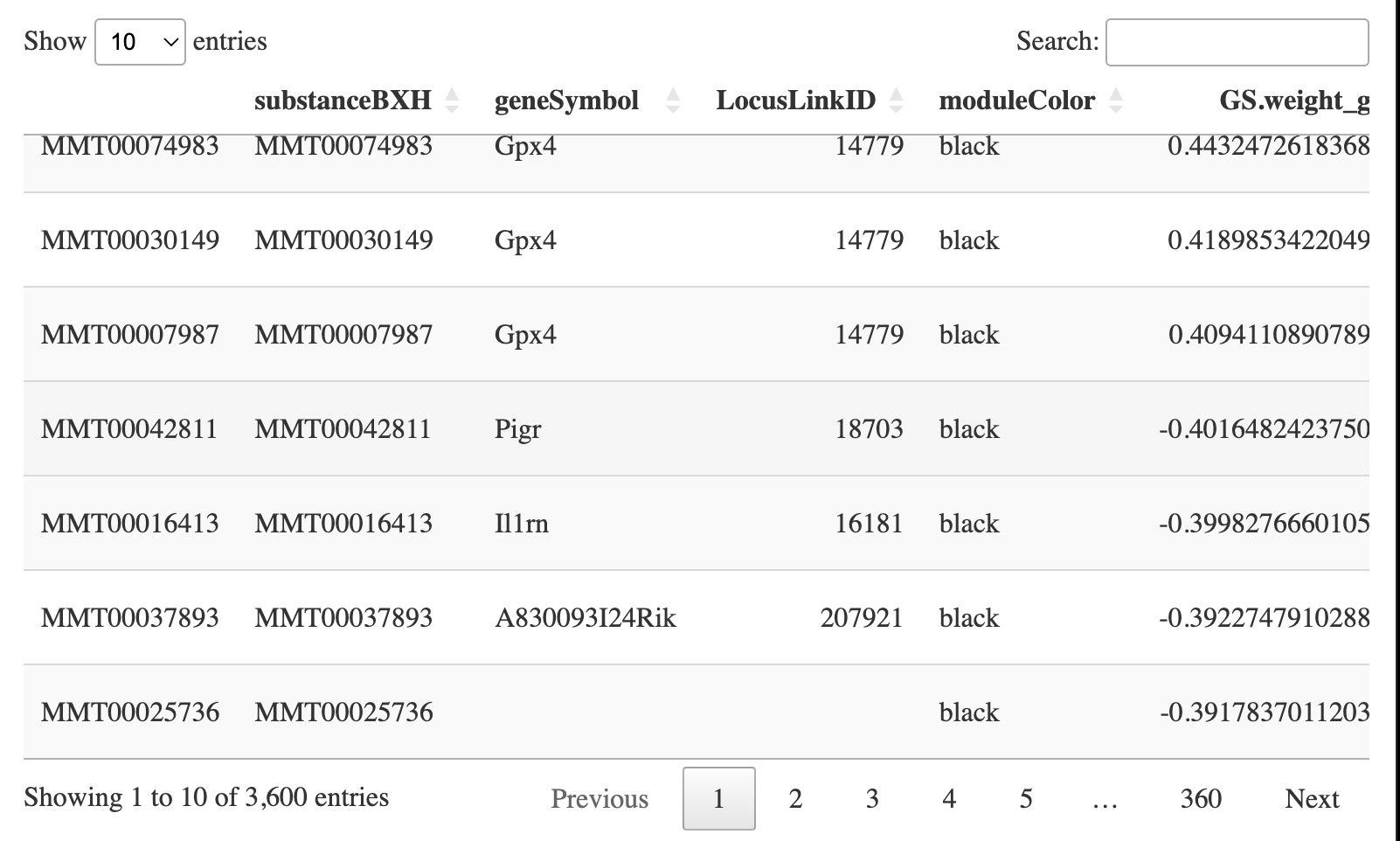
6如何引用
Langfelder, P., Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). https://doi.org/10.1186/1471-2105-9-559

???? ???? ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
???? ???? Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
???? ???? scRNA-seq | 吐血整理的单细胞入门教程
???? ???? NetworkD3 | 让我们一起画个动态的桑基图吧~
???? ???? RColorBrewer | 再多的配色也能轻松搞定!~
???? ???? rms | 批量完成你的线性回归
???? ???? CMplot | 完美复刻Nature上的曼哈顿图
???? ???? Network | 高颜值动态网络可视化工具
???? ???? boxjitter | 完美复刻Nature上的高颜值统计图
???? ???? linkET | 完美解决ggcor安装失败方案(附教程)
???? ......
本文由 mdnice 多平台发布
最后
以上就是勤奋香水最近收集整理的关于WGCNA | 值得你深入学习的生信分析方法!~(网状分析-第三步-模块与特征分析)的全部内容,更多相关WGCNA内容请搜索靠谱客的其他文章。

![Profiler EditorOnly [Loading.CheckConsistency [Editor Onlye]]](https://www.shuijiaxian.com/files_image/reation/bcimg12.png)






发表评论 取消回复