本文目标是:通过分析单细胞的数据,根据已有的细胞分型,去看我们感兴趣的基因集在这些细胞类型中的富集情况。单细胞数据和bulk数据会有些不同,可能一些具体的技巧需要注意一下。
1。切换到R4环境,加载RDS数据。
conda activate r4
R #进入到R
data<-readRDS("merge_obj.rds") # 加载原始数据
library(Seurat)#加载Seurat包
levels(data) #查看数据集的level
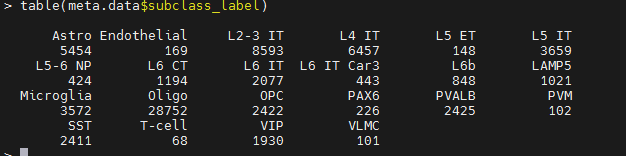
[1] "L5 IT" "L4 IT" "L2-3 IT" "L6b" "L6 IT Car3"
[6] "L6 IT" "L6 CT" "L5 ET" "L5-6 NP" "PVALB"
[11] "LAMP5" "SST" "VIP" "PAX6" "Oligo"
[16] "OPC" "Astro" "Microglia" "Endothelial" "T-cell"
[21] "PVM" "VLMC"
#这个数据集是已经处理好的数据,已知细胞分型的标签。
2。按照细胞类型,计算每一种细胞类型该基因表达值的均值
#首先要使得样本之间可以相互比较,需要对数据首先进行归一化处理
#首先,提取meta.data数据中的细胞类型的分类信息
meta.data<-data@meta.data #对SeuratObject对象不是很清楚啊 #那就一点点的去磨
table(meta.data$subclass_label)
#这个要快点做,感觉要计算好久。
#我想知道这个cellSubtypes参数是什么?看一下源代码#有顺序的label变量
cellSubtypes<-meta.data$subclass_label #这个为每一个细胞对应的label信息
subtypeOrder<-c("L2-3 IT","L4 IT","L5 ET","L5 IT","L5-6 NP","L6 CT","L6 IT Car3","L6b","LAMP5","PAX6","PVALB","PVM","SST","VIP","VLMC","Astro","Endothelial","Microglia","Oligo","OPC","T-cell")
#上述都准备好了之后
#选择归一化后的数据进行这一步的分析
#这里就遇到了一个问题:到底是使用标准化后的数据,还是归一化之后的数据呢?对于这个问题,请看下面的知识窗。这也是我不知道的,或者现在还没有弄明白的地方。
data@assays$RNA@data[1:5,1:5]
5 x 5 sparse Matrix of class "dgCMatrix"
B5_AAACAGCCAAATATCC-1 B5_AAACAGCCAGGAATCG-1 B5_AAACCAACAGGCCAAA-1
AL627309.1 . . .
AL627309.5 . . .
AL627309.4 . . .
AP006222.2 . . .
AC114498.1 . . .
B5_AAACCGAAGCCACATG-1 B5_AAACCGCGTACCGTTT-1
AL627309.1 . .
AL627309.5 . .
AL627309.4 . .
AP006222.2 0.3099514 .
AC114498.1 . .
> data@assays$RNA@scale.data
<0 x 0 matrix>
#从中可以看出,师兄的这套数据还没有进行scale处理(我觉得这种称呼很让人生气,觉得scale命名是画到同一个量纲中的方法)
#我想画一个boxplot图,看看输出是什么?
#pdf("boxplot.pdf")
#boxplot(data@assays$RNA@data[,1:5]) #很遗憾,由于数据格式转换的问题,有一些数据我还是很陌生
#dev.off()
#上面这个boxplot画不了
mean(data@assays$RNA@data[,1])
[1] 0.135663
mean(data@assays$RNA@data[,2])
[1] 0.1310064
mean(data@assays$RNA@data[,3])
[1] 0.1309113
mean(data@assays$RNA@data[,4])
[1] 0.1424853
#但是通过求mean值,可以推测数据是处于我们认为的比较理想的可比较的状态的。
#使用标准化之后的数据进行下一步的分析
dat<-data@assays$RNA@data
#计算每一种细胞类型的均值
cellTypeMean <- t(apply(dat, 1, function(v){
tapply(v, droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean)
}))
知识窗(标准化和归一化之间的区别和选择):
待补充。
这里要寻求完全的理解,需要对数据进行
报错1:
 看到一处,把稀疏矩阵转换为普通矩阵的方法:
看到一处,把稀疏矩阵转换为普通矩阵的方法:
参考链接:https://blog.csdn.net/u012110870/article/details/102804616
(真的,要学的有很多啊)
as_matrix <- function(mat){
tmp <- matrix(data=0L, nrow = mat@Dim[1], ncol = mat@Dim[2])
row_pos <- mat@i+1
col_pos <- findInterval(seq(mat@x)-1,mat@p[-1])+1
val <- mat@x
for (i in seq_along(val)){
tmp[row_pos[i],col_pos[i]] <- val[i]
}
row.names(tmp) <- mat@Dimnames[[1]]
colnames(tmp) <- mat@Dimnames[[2]]
return(tmp)
}
#使用上述代码,将稀疏矩阵转换为普通矩阵,进行加和处理。
#但是后来试了一下,好像不用转换也能计算。
tapply(dat[1,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean)
L2-3 IT L4 IT L5 ET L5 IT L5-6 NP L6 CT
0.020934889 0.030879634 0.028600512 0.031351582 0.026155217 0.029208913
L6 IT Car3 L6b LAMP5 PAX6 PVALB PVM
0.017774997 0.020983053 0.011340905 0.010886903 0.012902577 0.007187926
SST VIP VLMC Astro Endothelial Microglia
0.013860558 0.010441864 0.008101197 0.010201497 0.008115806 0.042921126
Oligo OPC T-cell
0.005094527 0.005365111 0.035109768
#不知道如果用最原始的for循环,是否可以实现这种功能
n<-dim(dat)[1]
feature<-as.data.frame(tapply(dat[1,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(feature)[1]<-rownames(dat)[1]
for(i in 2:n){
features<-as.data.frame(tapply(dat[i,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(features)[1]<-rownames(dat)[i]
feature<-cbind(feature,features)
}
#直接进行30000次循环即可,具体运行时间未知
#如果一秒得到计算结果,两秒合并数据框的话,那么30000次循环需要多少秒
#好吧急也急不来,需要8小时才能运行完成
到这里汇总一下,所有的有效代码(除去错误尝试的)。
data<-readRDS("merge_obj.rds")
meta.data<-data@meta.data
cellSubtypes<-meta.data$subclass_label
subtypeOrder<-c("L2-3 IT","L4 IT","L5 ET","L5 IT","L5-6 NP","L6 CT","L6 IT Car3","L6b","LAMP5","PAX6","PVALB","PVM","SST","VIP","VLMC","Astro","Endothelial","Microglia","Oligo","OPC","T-cell")
dat<-data@assays$RNA@data
n<-dim(dat)[1]
feature<-as.data.frame(tapply(dat[1,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(feature)[1]<-rownames(dat)[1]
for(i in 2:n){
features<-as.data.frame(tapply(dat[i,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(features)[1]<-rownames(dat)[i]
feature<-cbind(feature,features)
}
由于计算成本很大,本来想要挂一晚上,今天收结果的,很不幸的是,由于写代码的时候,一个变量的差别,导致最后输出的变量是计算后的中间变量,毫无意义。也算是给自己一个教训,要特别细心,注意细节,要不然特别容易犯错。
- #PBS文件
#PBS -N zhulab
#PBS -q batch
#PBS -o /home/xxzhang/workplace/cell_specific/celltype.out
#PBS -e /home/xxzhang/workplace/cell_specific/celltype.err
#PBS -l nodes=1:ppn=8
cd /home/xxzhang/workplace/cell_specific/
source activate r4 #这个source是我昨天学到的
Rscript cellType.r
- #Rscript
library(Seurat)
setwd("/home/xxzhang/workplace/cell_specific/")
data<-readRDS("merge_obj.rds")
meta.data<-data@meta.data
cellSubtypes<-meta.data$subclass_label
subtypeOrder<-c("L2-3 IT","L4 IT","L5 ET","L5 IT","L5-6 NP","L6 CT","L6 IT Car3","L6b","LAMP5","PAX6","PVALB","PVM","SST","VIP","VLMC","Astro","Endothelial","Microglia","Oligo","OPC","T-cell")
dat<-data@assays$RNA@data
n<-dim(dat)[1]
feature<-as.data.frame(tapply(dat[1,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(feature)[1]<-rownames(dat)[1]
for(i in 2:n){
features<-as.data.frame(tapply(dat[i,], droplevels(factor(cellSubtypes, levels=subtypeOrder)), mean))
colnames(features)[1]<-rownames(dat)[i]
feature<-cbind(feature,features) #目测是这里出现了问题
}
write.csv(feature,"output.csv")#实际上是这里
好的,还真是有很多经验和教训啊。快点给自己弄点吃的,然后去楼下自习。
提高代码的稳健性,还有一点就是增加与用户之间的互动,如输出确认。还有增加循环遍历的进度条;这些都是一个好的程序员的基本素养。
计算完成,保存为output.csv文件。
3。 计算PEM值。
data2<-t(data)
colnames(data2)<-data2[1,]
rownames(data2)<-rownames(dat)
cellTypeMean<-data2
cellTypeMean<-apply(cellTypeMean,2,as.numeric)
cellTypeMean2 <- cellTypeMean + 0.01
cellTypePEM <- t(apply(cellTypeMean2, 1, function(v) {
log10(v/cellTypeS*sum(cellTypeS, na.rm=TRUE)/sum(v, na.rm=TRUE))
}))
rownames(cellTypePEM)<-rownames(dat)
4。使用KS test评估我们感兴趣的基因的富集情况。
interest<-read.table("protein_coding_gene_interest_uniq.txt")
head(interest)
genes<-interest$V1
P.score<-KS_p(genes,cellTypePEM)
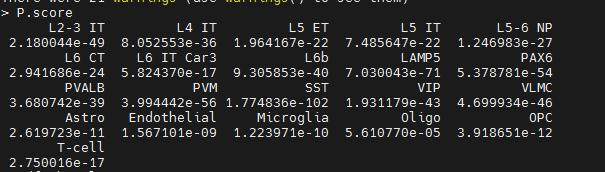 每一种细胞类型得到一个P值,P值越小,越富集。
每一种细胞类型得到一个P值,P值越小,越富集。
5。可视化
pdf("barplot3.pdf")
par(cex=0.5,las=2)
barplot(log(P.score),ylab = "log(p.value)",xlab = "cell type")
dev.off()
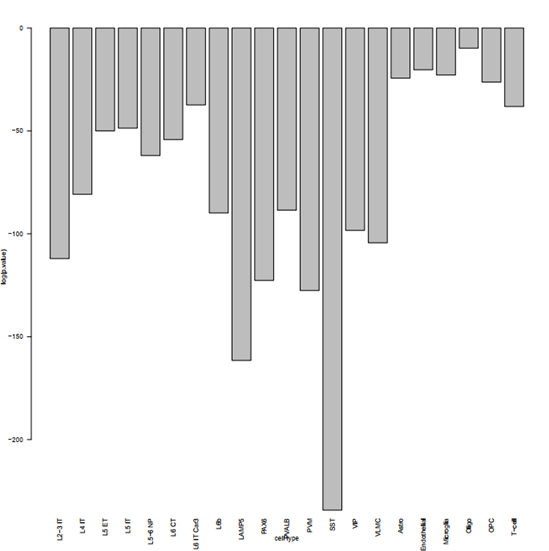
基本实现项目需求。
最后
以上就是合适黑夜最近收集整理的关于单细胞基础分析 | 基因细胞类型特异性富集分析的全部内容,更多相关单细胞基础分析内容请搜索靠谱客的其他文章。
本图文内容来源于网友提供,作为学习参考使用,或来自网络收集整理,版权属于原作者所有。




![Profiler EditorOnly [Loading.CheckConsistency [Editor Onlye]]](https://www.shuijiaxian.com/files_image/reation/bcimg12.png)


发表评论 取消回复