QTLseqr是一个用于BSA-seq的R包,它能够读取GATK输出结果进行过滤,最终使用SNP-index和G 统计值的方法进行定位。我们这次尝试使用这个R包来替代我们自己手撸代码来分析一批水稻BSA数据,文章的数据在如何使用BSA方法进行遗传定位(水稻篇)提供了下载链接。
首先是安装和加载R包,由于QTLseqr目前托管在GitHub上,因此需要用devtools才能安装。
devtools::install_github("bmansfeld/QTLseqr")
library("QTLseqr")
在读取数据上,QTLseqr提供了两个函数,importFromGATK和importFromTable,而且这两个函数都没有设置亲本样本名的参数,只有highBulk和lowBulk设置两个混池的样本名。这就很尴尬了,因为我发现直接用importFromGATK读取有4个样本的VCF文件会报错。
不过没关系,我们先将数据预处理成importFromTable能够识别的格式,就可以进行读取了。
我们先用vcfR提取所需要的信息列,包括AD和GT,前者是等位基因深度,后者是基因型
library(vcfR)
vcf <- read.vcfR("04-variant-filter/snps.vcf.gz")
chrom <- getCHROM(vcf)
pos <- getPOS(vcf)
ref <- getREF(vcf)
alt <- getALT(vcf)
ad <- extract.gt(vcf, "AD")
ref_split <- masplit(ad, record = 1, sort = 0)
alt_split <- masplit(ad, record = 2, sort = 0)
gt <- extract.gt(vcf, "GT")
补充下masplit函数的用法,这函数来自于vcfR, 他的作用就是对数据进行分列,注意, 它返回的是矩阵中每个元素拆分后其中一个值,主要参数为
- myMat 输入矩阵
- delim = “,” 矩阵里元素的分隔符号,
- count = 0L: 0或1,也就是False或True,统计每个元素有多少个子元素,例如’1,2,3’ 会返回3, ‘1,2’ 返回2, 一般用不到
- record = 1L: 返回分隔之后的第几个元素,
- sort = 1L: 是否排序
- decreasing = 1L: 是否降序
之后构建数据框, 过滤亲本杂合的位点,并保存到本地
df <- data.frame(CHROM = chrom,
POS = pos,
REF = ref,
ALT = alt,
AD_REF.SRR6327817 = ref_split[,3],
AD_ALT.SRR6327817 = alt_split[,3],
AD_REF.SRR6327818 = ref_split[,4],
AD_ALT.SRR6327818 = alt_split[,4]
)
mask <- which(gt[,"SRR6327815"] != "0/1" & gt[,"SRR6327816"] != "0/1")
df <- df[mask,]
write.table(df, file = "rice.tsv", sep = "t", row.names = F, quote = F)
之后用importFromTable读取,并过滤掉SNPindex为NaN的行
df <- importFromTable("rice.tsv",
highBulk = "SRR6327817",
lowBulk = "SRR6327818",
chromList = paste0("chr", formatC(1:12, width = 2, flag=0)),
sep = "t")
df <- subset(df, !is.na(SNPindex.LOW) & !is.na(SNPindex.HIGH))
分别运行两种BSA的分析方法,windowSize指的是窗口大小
df <- runGprimeAnalysis(SNPset = df,
windowSize = 1e6,
outlierFilter = "deltaSNP")
df <- runQTLseqAnalysis(SNPset = df,
windowSize = 1e6,
popStruc = "RIL",
bulkSize = c(20,20))
最后我们可视化结果
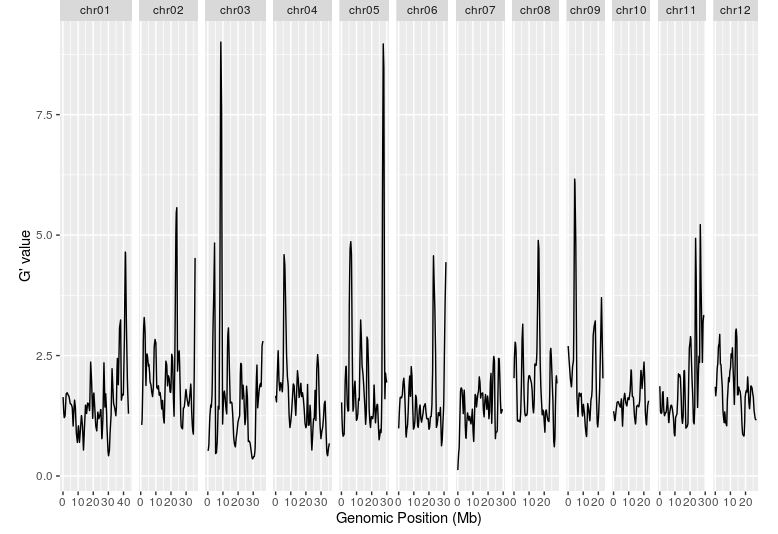
plotQTLStats(
SNPset = df,
var = "Gprime",
plotThreshold = TRUE,
q = 0.01
)
plotQTLStats(
SNPset = df,
var = "deltaSNP",
plotIntervals = TRUE)
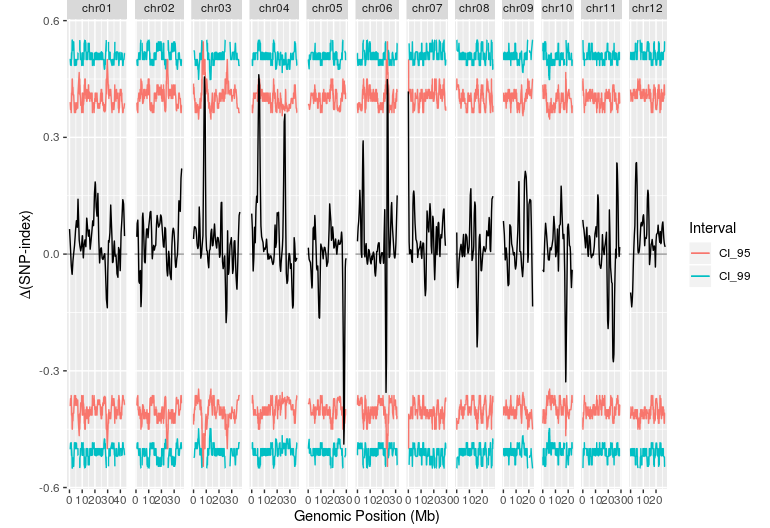
结果和文章比较吻合,在6号染色体有一个非常明显的峰。
QTLseqr这个R包整体的表现还是可以的,就是对输入有一定的要求,如果直接提供VCF文件需要保证你的VCF文件只有两个样本。也就是你需要先单独对亲本做变异检测,然后用这些位点对两个混池变异检测结果进行过滤,不然这两个亲本的简直就体现不出来了。
版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。

最后
以上就是称心滑板最近收集整理的关于使用QTLseqr进行BSA-seq分析的全部内容,更多相关使用QTLseqr进行BSA-seq分析内容请搜索靠谱客的其他文章。







发表评论 取消回复