甲基化芯片学习
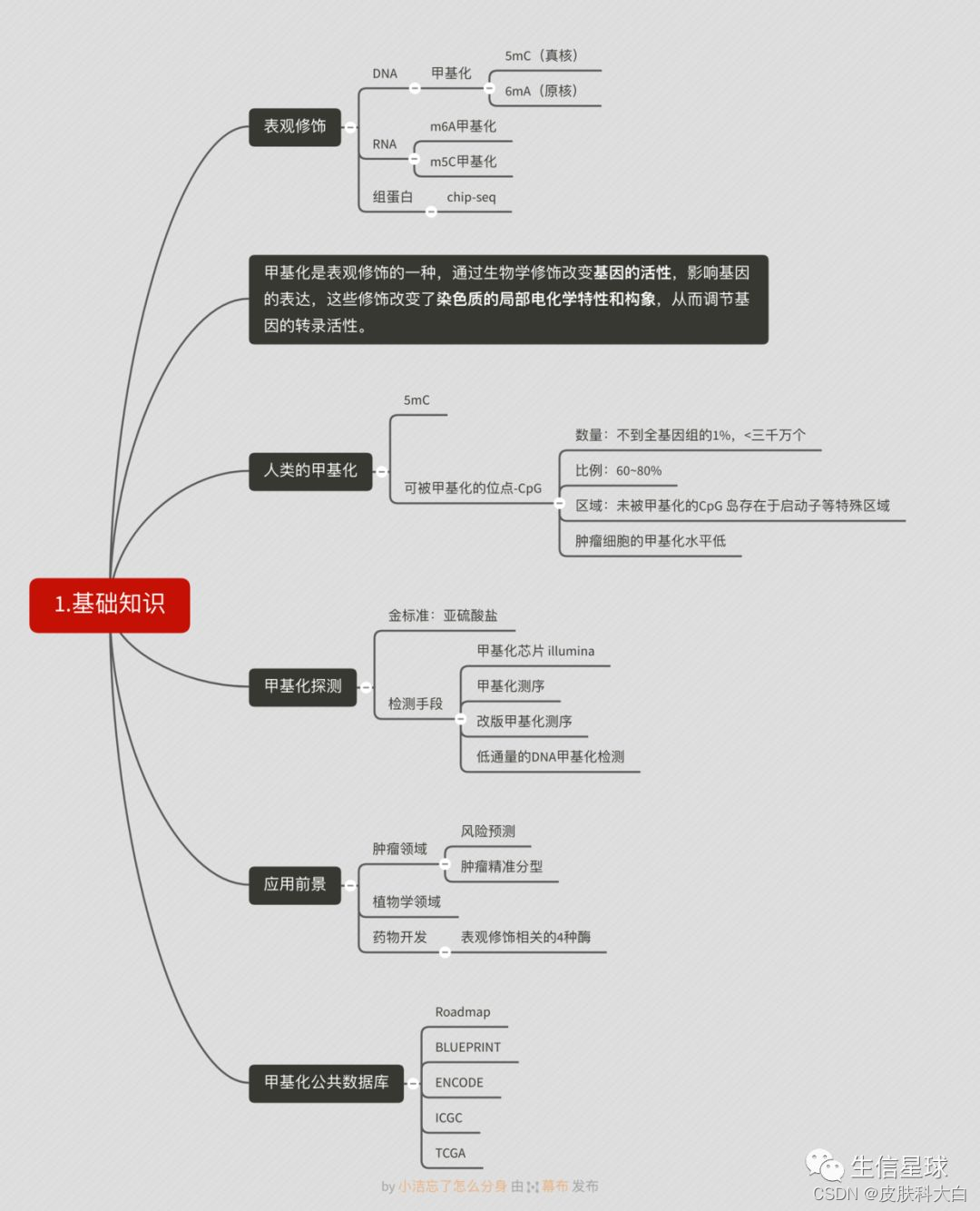
第二部分
,甲基化芯片分析的一般流程,包括了甲基化信号值的概念,可以做什么分析等,参考资料:
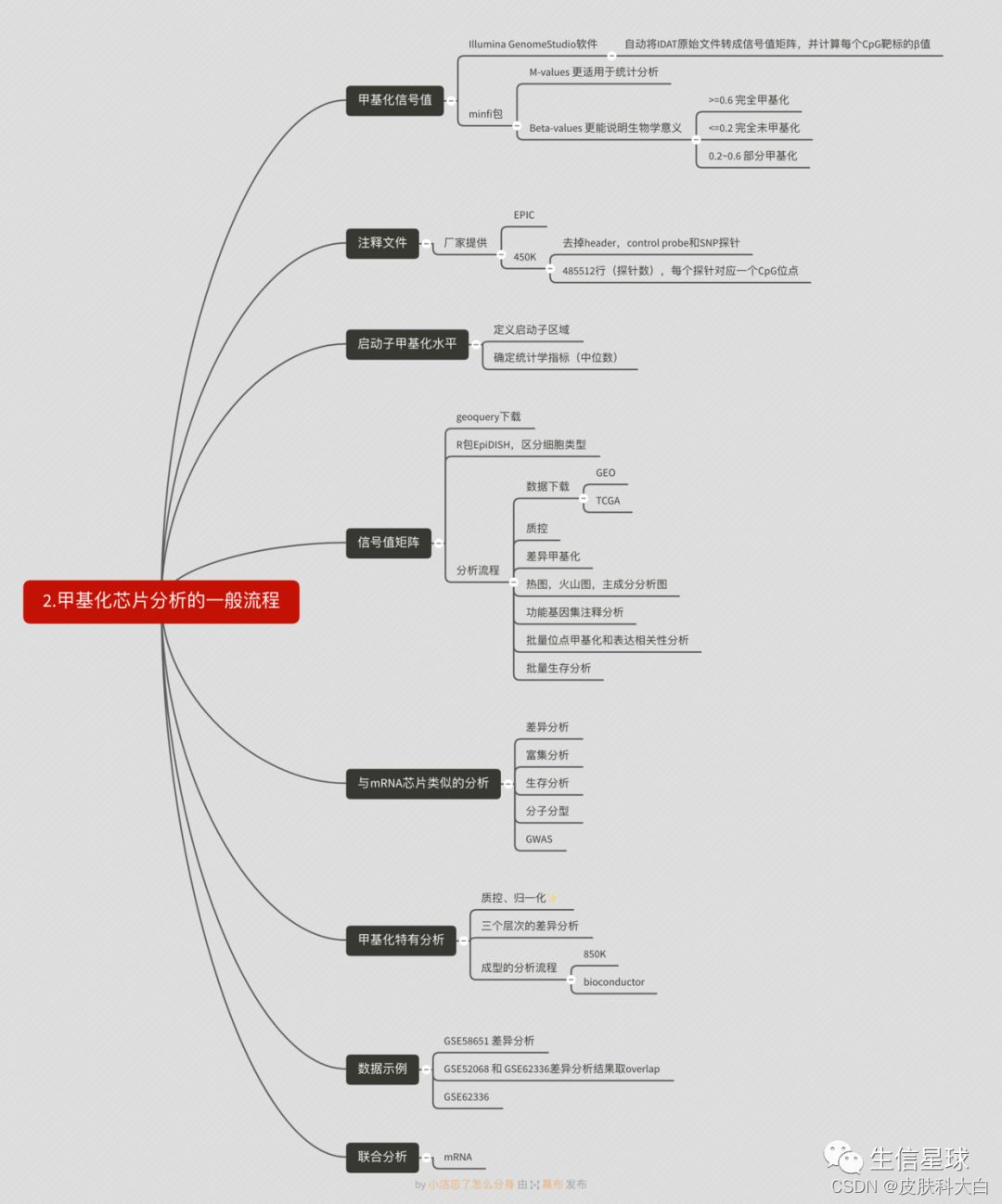
第三部分 Bioconductor的代码流程
主要是使用了R包minfi做质控和标准化。
中文导读:Bioconductor的DNA甲基化芯片分析流程
原文网址:http://www.bioconductor.org/help/workflows/methylationArrayAnalysis/
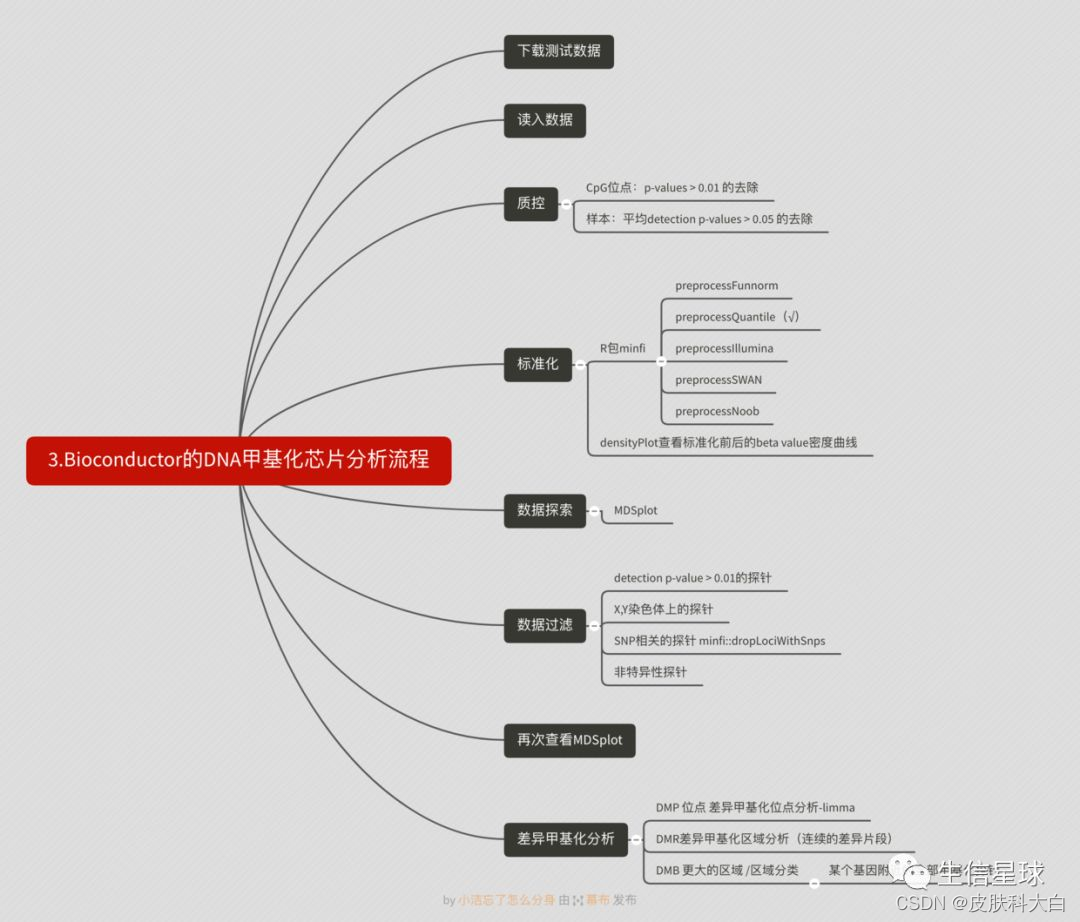
最后标准流程,包括数据下载、质控及差异分析、富集分析,香!
数据下载:甲基化芯片数据下载如何读入到R里面
质控:甲基化芯片数据的一些质控指标
一次偶然的搜索中发现biocondutor有个甲基化芯片的分析流程,刚好可以学习下,写的真的很棒。
Bioconductor的DNA methylation workflow可以在http://www.bioconductor.org/help/workflows/methylationArrayAnalysis/中查看,教程开头先对DNA甲基化芯片及其原理做了简单的介绍,包括一些常见的术语:比如β value和M value,后面就正式进入DNA甲基化的分析方法的讲解了。
下载测试数据
测试数据是放在methylationArrayAnalysis中的,安装官网的方法安装下即可(PS.如果安装时卡住,可以用Rstudio的Tools来安装)
source("http://bioconductor.org/workflows.R")
workflowInstall("methylationArrayAnalysis")
安装后,比如我R包是默认安装的R文件夹中的,所以我的数据路径是E:/R-3.4.1/library/methylationArrayAnalysis/extdata/,那么可以列出该目录下有哪些文件看看
list.files("E:/R-3.4.1/library/methylationArrayAnalysis/extdata/", recursive = TRUE)
测试数据总共有两套数据(GSE49667和GSE51180),前者总共包括10个样本,作为主要的测试数据;后者则只用了其一个样本,作为测试数据中的异常样本。样本分组信息:从3个体(M28, M29, M30)中取4种不同的T-cell types(naive, rTreg, act_naive, act_rTreg),act_naive、act_rTreg是指对应经anti-CD3、anti-CD28 antibodies处理的naive、rTreg,以上分组信息也可以看数据目录下的SampleSheet.csv文件
作者提了最近几年常见的DNA甲基化芯片分析的软件,如minfi,methylumi,wateRmelon,missMethyl,ChAMP和charm,最后还提到了用limma来做差异甲基化位点分析
读入数据
先按照教程加载其所会用到的R包
library(limma)
library(minfi)
library(IlluminaHumanMethylation450kanno.ilmn12.hg19)
library(IlluminaHumanMethylation450kmanifest)
library(RColorBrewer)
library(missMethyl)
library(matrixStats)
library(minfiData)
library(Gviz)
library(DMRcate)
library(stringr)
其中minfi,IlluminaHumanMethylation450kanno.ilmn12.hg19,IlluminaHumanMethylation450kmanifest,missMethyl,DMRcate是DNA甲基化分析专用R包,RColorBrewer,Gviz是用于可视化的R包,stringr,matrixStats是用于一些数据处理的,limma则是用于差异甲基化分析的
用minfi包的read.metharray.sheet函数读入IDAT格式的数据,其中SampleSheet.csv文件是必须的,不然最后数据集中会缺少样本信息;其产生的targets向量中包含了样本信息外,还有IDAT格式数据的所在路径,方便read.metharray.exp函数读入数据
targets <- read.metharray.sheet("E:/R-3.4.1/library/methylationArrayAnalysis/extdata", pattern="SampleSheet.csv")
rgSet <- read.metharray.exp(targets=targets)
质控
以detection p-values评估每样本中的每个CpG位点的质量,当p-values > 0.01时则说明该探针信号数据质量较低,需要去除
detP <- detectionP(rgSet)
将平均detection p-values > 0.05的样本认为是低质量的样本;从结果中可以看出最后一个样本的平均detection p-values远远大于0.05,因此从rgSet中去除(从rgSet的结构中看出,其数据分布是按照每列一个样本来的)
keep <- colMeans(detP) < 0.05
keep
rgSet <- rgSet[,keep]
标准化
现在有很多用于DNA甲基化芯片数据的标准化方法,其中一些方法已经整合在minfi包中了;现在为止没有一种标准化方法被认为是最好的方法,但是一些标准化方法在某些情况下是比较好的选择,作者总结了如下几点:
1.对于用于cancer/normal or vastly different tissue types的差异比较的话,可以用preprocessFunnorm函数(Functional normalization)
2.对于不需要进行差异比较的单样本数据,如单个组织样本,那么可以用preprocessQuantile函数
3.更多的标准化方法的比较,作者推荐看A Data-Driven Approach to Guide the Choice of an Appropriate Normalization Method
除了上述提的2个标准化方法外,minfi包还支持preprocessIllumina(类似于Illumina的Genome Studio的标准化方法),preprocessSWAN(Subset-quantile within array normalisation)以及preprocessNoob(normal-exponential out-of-band)
作者比较了这些标准化方法后,发现对于这次的测试数据,这几种标准化结果都大体上相似的,所以作者最终选择了用preprocessQuantile,标准化为rgSet从RGChannelSet对象变成了mSetSq的GenomicRatioSet对象,表示从探针的信号强度转化到了beta value/M value,并且已mapping to Genome
mSetSq <- preprocessQuantile(rgSet)
我们也可以不做任何标准化处理,只为获得GenomicRatioSet对象用于下游分析
mSetRaw <- preprocessRaw(rgSet)
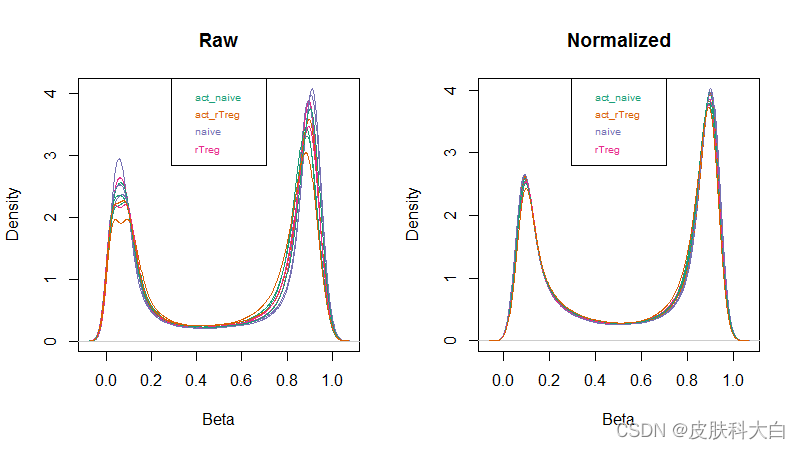
然后用minfi包内置的densityPlot函数,看看标准化前后的beta value的密度曲线
targets <- targets[keep,] ##在样本信息中也去掉异常样本
par(mfrow=c(1,2))
densityPlot(rgSet, sampGroups=targets$Sample_Group,main="Raw", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"), cex = 0.6)
densityPlot(getBeta(mSetSq), sampGroups=targets$Sample_Group, main="Normalized", legend=FALSE)
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"), cex = 0.6)
Data exploration
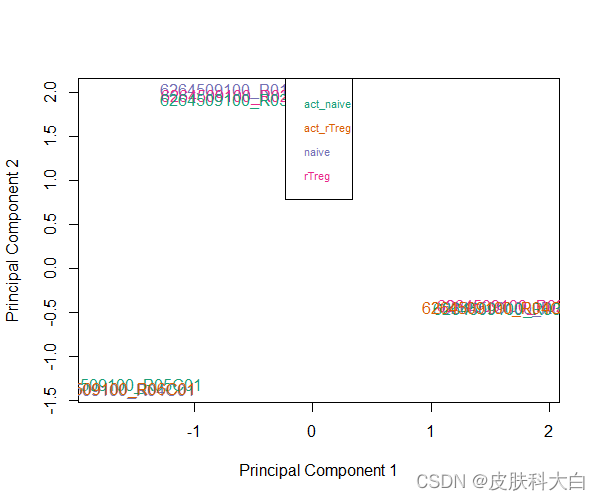
作者建议可以先做个主成分分析,如Multi-dimensional scaling (MDS) plots来看下样本分布情况,可以根据情况自行选择target向量中的分组,如看下targets$Sample_Group下的MDS plot,PS.这里是用M value来作图的
plotMDS(getM(mSetSq), top=1000, gene.selection="common", col=pal[factor(targets$Sample_Group)])
legend("top", legend=levels(factor(targets$Sample_Group)), text.col=pal, bg="white", cex=0.7)
数据过滤
一些质量较差的探针需要在下游差异分析之前去除,之前的质控作者是用detection p-value去除了异常样本,在这里则需要去除在一些样本中质量较差的探针
因为前面质控时获得的detP中的探针顺序已经和mSetSq中的探针不一致了,所以先将前者的顺序调整为和后者一致
detP <- detP[match(featureNames(mSetSq),rownames(detP)),]
- 作者以remove any probes that have failed in one or more samples为标准去除不满足detection p-value阈值(0.01)的探针,也就是说,一个探针只要detection p-value > 0.01就去除
keep <- rowSums(detP < 0.01) == ncol(mSetSq)
table(keep)
mSetSqFlt <- mSetSq[keep,]
- 过滤掉X,Y染色体上的探针(一般数据都要做这一步,只要还需进行下游差异分析的话),但这个测试数据都是男性样本,所以就没必要去除了,但作者还是给出了该怎么去除在X,Y染色体上探针的代码,原理就是根据ann450k = getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19)这个注释文件的数据集,里面标注了那些探针出现在哪个染色体上的信息
keep <- !(featureNames(mSetSqFlt) %in% ann450k`$Name[ann450k$`chr %in% c("chrX","chrY")])
table(keep)
mSetSqFlt <- mSetSqFlt[keep,]
- 过滤掉所有SNP相关的探针,作者建议用minfi包的dropLociWithSnps函数,除了去除默认的SNPs位点外,还可以去除那些minor allele frequencies高于阈值的位点;注:该函数是默认去除SNPs出现在CpG or single base extension (SBE) site的探针
mSetSqFlt <- dropLociWithSnps(mSetSqFlt, snps = c("CpG", "SBE"))
mSetSqFlt
- 去除cross-reactive的探针,也就是multi-hit探针,即映射到多个位置的;这探针list是Chen et al.(2013)发表的,所以只要出现在这list上面的探针都可以去除了
dataDirectory <- "E:/R-3.4.1/library/methylationArrayAnalysis/extdata/"
xReactiveProbes <- read.csv(file=paste(dataDirectory, "48639-non-specific-probes-Illumina450k.csv", sep="/"), stringsAsFactors=FALSE)
keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID)
mSetSqFlt <- mSetSqFlt[keep,]
mSetSqFlt
做完上述过滤和标准化后,作者建议再看下MDS plot,看看样本之间的联系是否发生了改变。对这个测试数据而言,第一个主成分上样本不像之前那么集中了,作者怀疑是由于去除了SNP关联的探针的缘故
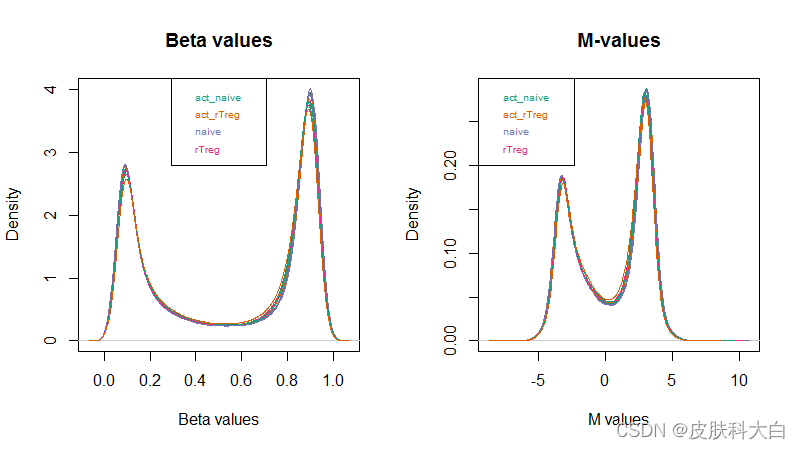
作者使用minfi包的getM和getBeta来分别计算M-values和Beta-values,作者认为M-values具有更好的统计特性,更适合用于进行下游的统计分析(差异分析等);而Beta-values更加容易解释,更能说明生物学上的意义
mVals <- getM(mSetSqFlt)
bVals <- getBeta(mSetSqFlt)
并对两者分别做了密度分布图
par(mfrow=c(1,2))
densityPlot(bVals, sampGroups=targets$Sample_Group, main="Beta values", legend=FALSE, xlab="Beta values")
legend("top", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"))
densityPlot(mVals, sampGroups=targets$Sample_Group, main="M-values", legend=FALSE, xlab="M values")
legend("topleft", legend = levels(factor(targets$Sample_Group)), text.col=brewer.pal(8,"Dark2"))
差异甲基化分析
作者选了M-values作为后续分析的矩阵,对于差异甲基化位点的分析,作者没有用minfi包的dmpFinder函数,而是用了limma包的linear model来分析。所以按照limma的分析思路:
- 构建分组矩阵
cellType <- factor(targets$Sample_Group)
individual <- factor(targets$Sample_Source)
design <- model.matrix(~0+cellType+individual, data=targets)
colnames(design) <- c(levels(cellType),levels(individual)[-1])
- 构建两两比较矩阵
contMatrix <- makeContrasts(naive-rTreg,
naive-act_naive,
rTreg-act_rTreg,
act_naive-act_rTreg,
levels=design)
- 差异分析
fit <- lmFit(mVals, design)
fit2 <- contrasts.fit(fit, contMatrix)
fit2 <- eBayes(fit2)
- 将位点信息和limma差异分析的结果合并在一起,这里以naive - rTreg为例
ann450kSub <- ann450k[match(rownames(mVals),ann450k$Name), c(1:4,12:19,24:ncol(ann450k))]
DMPs <- topTable(fit2, num=Inf, coef=1, genelist=ann450kSub)
head(DMPs)
除了差异甲基化位点分析,作者还提了怎么进行DMR(Differential Methylation Regions)分析;作者总结了几个可用来DMR分析的bioconductor包,如:minfi包的bumphunter函数,DMRcate包的dmrcate函数。由于作者觉得bumphunter函数运算速度过慢,除非使用多线程并行运算(如doParallel包),所以选择用dmrcate函数;并且由于这个函数也是基于limma,所以可以直接使用上面的design和contMatrix
- 先用DMRcate包的cpg.annotate函数做单个甲基化位点的差异分析以及注释
myAnnotation <- cpg.annotate(object = mVals, datatype = "array", what = "M",
analysis.type = "differential", design = design,
contrasts = TRUE, cont.matrix = contMatrix,
coef = "naive - rTreg", arraytype = "450K")
- 接着用dmrcate函数在上面单个甲基化位点分析的基础上进行合并,从而识别出DMR(Differential Methylation Regions)
DMRs <- dmrcate(myAnnotation, lambda=1000, C=2)
head(DMRs$results)
- 用DMR.plot函数进行可视化,看看这些DMRs在基因组上的展现形式
data(dmrcatedata)
results.ranges <- extractRanges(DMRs, genome = "hg19")
# set up the grouping variables and colours
groups <- pal[1:length(unique(targets$Sample_Group))]
names(groups) <- levels(factor(targets$Sample_Group))
cols <- groups[as.character(factor(targets$Sample_Group))]
samps <- 1:nrow(targets)
# draw the plot for the top DMR
par(mfrow=c(1,1))
DMR.plot(ranges=results.ranges, dmr=1, CpGs=bVals, phen.col=cols, what = "Beta",
arraytype = "450K", pch=16, toscale=TRUE, plotmedians=TRUE,
genome="hg19", samps=samps)
Summary
我这只介绍了这篇Bioconductor workflow for analysing methylation array的部分内容,后续还有讲如何用Gviz包定制化展示甲基化数据,如何用missMethyl包做后续的GO/KEGG分析,还以一个肿瘤甲基化研究的文章为例,展示如何用甲基化来分析肿瘤样本的整个流程(整体上与上述的测试例子类似,但也有点不同)。总之,是一篇值得从头到尾好好看的DNA甲基化分析流程的文章!
一个甲基化芯片信号值矩阵差异分析的标准代码
学完这些对甲基化芯片有了一定的了解,还需要多看些文献,加油!
TCGA数据库的癌症甲基化芯片数据重分析
文献:Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma,下载TCGA数据库中口腔癌的甲基化芯片信号值矩阵,然后挑选有N-T配对的32个病人的数据进行差异分析,画火山图和热图。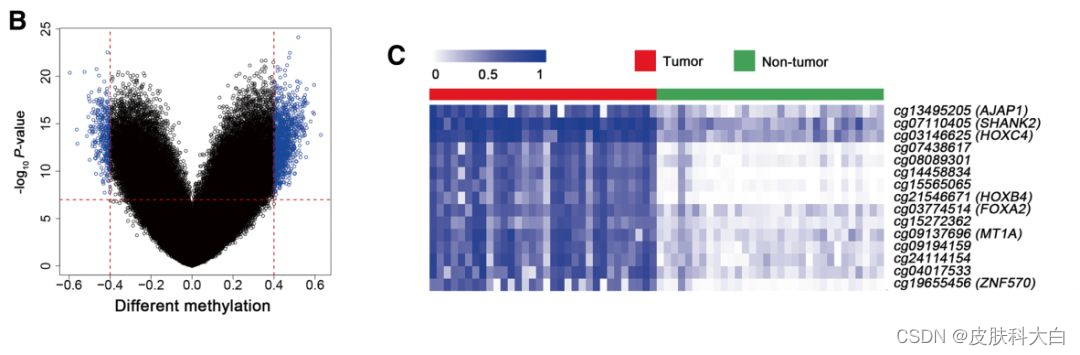
准备数据
- 从UCSC xena(https://xenabrowser.net/datapages/)中下载头颈癌HNSC的甲基化数据HumanMethylation450.gz(数据很大,如果下载失败,那应该是网络的问题),表型数据HNSC_clinicalMatrix.gz,生存数据HNSC_survival.txt.gz。
- 挑选有Normal-Tumor配对的32个OSCC病人
# UCSC的xena浏览器下载指定HNSC的phenotype数据
pd.all <- read.delim("./raw_data/HNSC_clinicalMatrix", header = T, stringsAsFactors = F)
colnames(pd.all)
pd <- pd.all[,c("sampleID","anatomic_neoplasm_subdivision","sample_type")]
table(pd$anatomic_neoplasm_subdivision)
# 挑出OSCC样本(参考文章methods)
oscc <- c("Oral Cavity","Oral Tongue","Buccal Mucosa","Lip","Alveolar Ridge","Hard Palate","Floor of mouth")
pd <- pd[pd$anatomic_neoplasm_subdivision %in% oscc,]
# 挑出normal-tumor配对样本
pd$patient <- substr(pd$sampleID,1,12)
pd <- pd[pd$patient %in% pd$patient[pd$sample_type=="Solid Tissue Normal"],]
rownames(pd) <- pd$sampleID
pd$sample_type <- ifelse(pd$sample_type=="Primary Tumor","Tumor","Normal")
# 保存配对样本(此时有48对)
write.table(pd$sampleID, file = "./raw_data/samples.txt",quote = F, row.names = F, col.names = F)
- 缩减甲基化信号矩阵:因为整个HNSC甲基化矩阵有580个样本,读进R会很慢,而且没有必要,所以先用linux命令根据sample.txt筛选出N-T配对的OSCC样本。
# 解压甲基化信号矩阵文件
$ gunzip HumanMethylation450.gz
# 文件内容是这样的
$ head -5 samples.txt
TCGA-CV-5436-01
TCGA-CV-5436-11
TCGA-CV-5442-01
TCGA-CV-5442-11
TCGA-CV-5966-01
$ cat HumanMethylation450 | cut -f 1-6 |head -5
sample TCGA-CN-6022-01 TCGA-CQ-5327-01 TCGA-CV-5435-11 TCGA-HD-7753-01 TCGA-CQ-5325-01
cg13332474 0.0220 0.0264 0.0307 0.0263 0.0218
cg00651829 0.0209 0.4514 0.0214 0.0231 0.0248
cg17027195 0.0443 0.2414 0.0330 0.0361 0.0416
cg09868354 0.0490 0.0546 0.0469 0.0754 0.0516
## 筛选N-T配对的OSCC样本
# 1.获得OSCC样本所在的列
$ cols=($(sed '1!d;s/t/n/g' HumanMethylation450 | grep -nf samples.txt | sed 's/:.*$//'))
# 2.筛选出OSCC样本的甲基化信号矩阵
$ cut -f 1$(printf ",%s" "${cols[@]}") HumanMethylation450 > OSCC_NT_methy450k.txt
载入数据为champ对象
## 安装ChAMP包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ChAMP") #有点漫长,可能会报错,把报错的包手动安装一下应该没什么问题
library("ChAMP")
## 读取具有N-T配对的OSCC甲基化信号矩阵
library(data.table)
a <- fread("./raw_data/OSCC_NT_methy450k.txt", data.table = F )
a[1:4,1:4]
rownames(a)=a[,1]
a=a[,-1]
beta=as.matrix(a)
# beta信号值矩阵里面不能有NA值
beta=impute.knn(beta)
betaData=beta$data
betaData=betaData+0.00001
sum(is.na(betaData))
#表型数据
pd <- pd[colnames(betaData),]
table(pd$patient) #此时有些normal样本没有配对的tumor
NT.s <- pd$patient[duplicated(pd$patient)] #得到32对样本,和文章一致了
pd <- pd[pd$patient %in% NT.s,]
betaData <- betaData[,pd$sampleID]
dim(betaData)
# 载入为ChAMP对象
library(ChAMP)
myLoad=champ.filter(beta = betaData ,pd = pd) #这一步已经自动完成了过滤
dim(myLoad$beta)
save(myLoad,file = './Rdata/step1-output.Rdata')
甲基化矩阵质控
rm(list = ls())
load('./Rdata/step1-output.Rdata')
# 数据归一化
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)
dim(myNorm)
QC.GUI(beta=myNorm,arraytype="450K") # 显示有NA值
num.na <- apply(myNorm,2,function(x)(sum(is.na(x))))
hist(num.na) # 看来是有样本有问题,个别样本校正后NA值过高
table(num.na) # 61个样本没有NA,3个样本含有超过250k个NA
myNorm <- myNorm[,which(num.na < 250000)] # 删掉异常样本
save(myNorm,file = './Rdata/step2-champ_myNorm.Rdata')
# 主成分分析
library("FactoMineR")
library("factoextra")
dat <- t(myNorm)
pd <- myLoad$pd[colnames(myNorm),] #去掉异常样本
group_list=pd$sample_type
table(group_list)
dat.pca <- PCA(dat, graph = FALSE)
fviz_pca_ind(dat.pca,
geom.ind = "point",
col.ind = group_list,
addEllipses = TRUE,
legend.title = "Groups")
# 热图
cg=names(tail(sort(apply(myNorm,1,sd)),1000))
library(pheatmap)
ac=data.frame(group=group_list)
rownames(ac)=colnames(myNorm)
pheatmap(myNorm[cg,],show_colnames =F,show_rownames = F,
annotation_col=ac,filename = 'heatmap_top1000_sd.png')
dev.off()
# 相关关系矩阵热图
# 组内的样本的相似性应该是要高于组间的!
pheatmap::pheatmap(cor(myNorm[cg,]),
annotation_col = ac,
show_rownames = F,
show_colnames = F)
# 去除异常样本
# 图中看出TCGA-CV-5971-01,TCGA-CV-6953-11,TCGA-CV-6955-11这3个样本有点异常
myNorm <- myNorm[,!(colnames(myNorm) %in% c("TCGA-CV-5971-01","TCGA-CV-6953-11","TCGA-CV-6955-11"))]
pd <- pd[colnames(myNorm),]
save(pd,myNorm,file = "./Rdata/filtered.Rdata")
差异分析
rm(list=ls())
load("./Rdata/filtered.Rdata")
# 差异分析
group_list <- pd$sample_type
myDMP <- champ.DMP(beta = myNorm,pheno=group_list)
head(myDMP[[1]])
save(myDMP,file = './Rdata/step3-output-myDMP.Rdata')
画图
根据差异分析结果画火山图,因为作者的差异分析用的是paired t test,和我的结果差异有点大,我自己选了差异甲基化的cutoff。
df_DMP <- myDMP$Tumor_to_Normal[,1:5]
logFC_cutoff <- 0.45
df_DMP$change <- ifelse(df_DMP$adj.P.Val < 10^-15 & abs(df_DMP$logFC) > logFC_cutoff,
ifelse(df_DMP$logFC > logFC_cutoff ,'UP','DOWN'),'NOT')
table(df_DMP$change)
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'nThe number of up gene is ',nrow(df_DMP[df_DMP$change =='UP',]) ,
'nThe number of down gene is ',nrow(df_DMP[df_DMP$change =='DOWN',]))
library(ggplot2)
g <- ggplot(data=df_DMP,
aes(x=logFC, y=-log10(adj.P.Val),
color=change)) +
geom_point(alpha=0.4, size=1) +
theme_set(theme_set(theme_bw(base_size=10)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=10,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red'))
print(g)
### 挑选影响生存的CpG位点,画热图
# 提取差异甲基化CpGs
choose_gene <- rownames(df_DMP[df_DMP$change != "NOT",])
choose_matrix <- myNorm[choose_gene,]
## 批量生存分析
# 载入生存信息
phe <- read.delim("./raw_data/HNSC_survival.txt.gz",header = T, stringsAsFactors = F)
phe <- phe[phe$sample %in% colnames(choose_matrix),]
phe <- phe[substr(phe$sample,14,15)=="01",]
choose_matrix <- choose_matrix[,phe$sample]
library(survival)
logrankP <- apply(choose_matrix, 1, function(x){
#x <- choose_matrix[1,]
phe$group <- ifelse(x>mean(x),"High","Low")
res <- coxph(Surv(OS.time, OS)~group, data=phe)
beta <- coef(res)
se <- sqrt(diag(vcov(res)))
p.val <- 1 - pchisq((beta/se)^2, 1)
})
names(logrankP) <- rownames(choose_matrix)
table(logrankP<0.05) #151个CpG位点
surv_gene <- names(sort(logrankP))[1:20] #挑了20个生存最显著的DMP,用于画图
# 热图
plot_matrix <- myNorm[surv_gene,]
annotation_col <- data.frame(Sample=myLoad$pd$sample_type ) #热图数据的bar
rownames(annotation_col) <- colnames(plot_matrix)
ann_colors = list(Sample = c(Normal="green", Tumor="red"))
library(pheatmap)
pheatmap(plot_matrix,show_colnames = F,annotation_col = annotation_col,
border_color=NA,
color = colorRampPalette(colors = c("white","navy"))(50),
annotation_colors = ann_colors)
最后
以上就是过时台灯最近收集整理的关于甲基化芯片学习的全部内容,更多相关甲基化芯片学习内容请搜索靠谱客的其他文章。








发表评论 取消回复