关于文献中二代测序数据下载(NCBI)的问题
现在二代测序用于生物学研究非常广泛,大部分文章的序列会上传到Sequence Read Archive(SRA)上,这东西也属于NCBI数据库中的吧,我理解是。
怎么从文献中下载这些序列呢?
首先在文章中找到作者提供的SRA号,或者SRP号。有的习惯写在材料方法中,有的习惯写在文章的末尾的Acknowlagements里面。本次的例子写在方法里面,如图。

L. Fernández Bidondo 的Detection of arbuscular mycorrhizal fungi …文章中的截图
打开NCBI官网
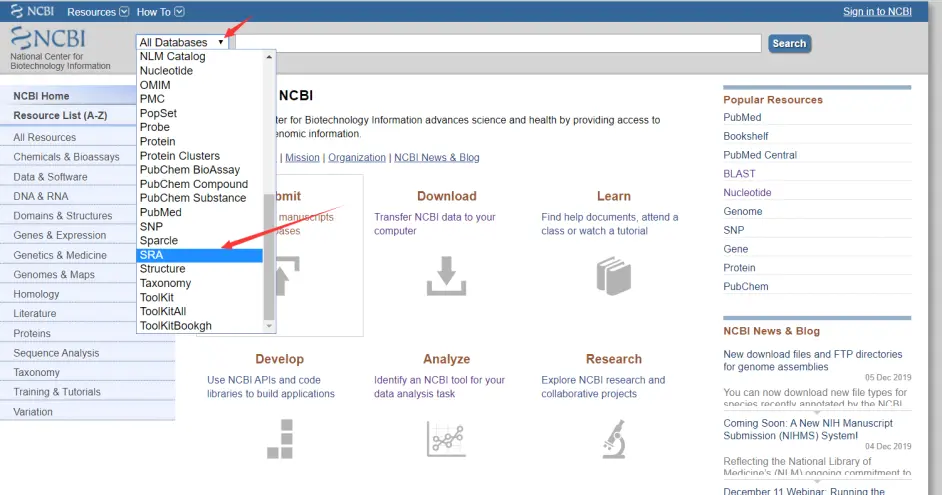
NCBI
选择SRA搜索文章中的SRA号
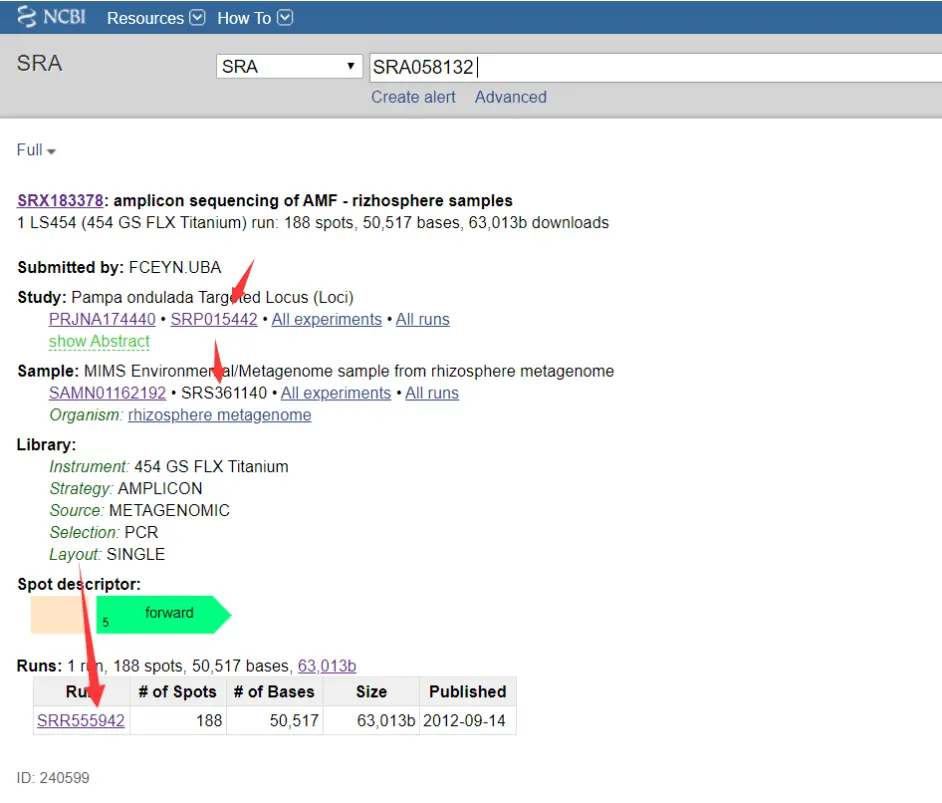
搜索结果
在 SRA 数据库中, 研究课题的检索号以前缀 DRP、ERP或SRP开头。
样本的检索号以前缀 DRS、ERS或SRS开头。
序列及其质量信息在SRA 数据库中以run为单元存储。run的检索号以前缀DRR、ERR或SRR开头。
我们下载序列,所以点击下面的SRR555942。
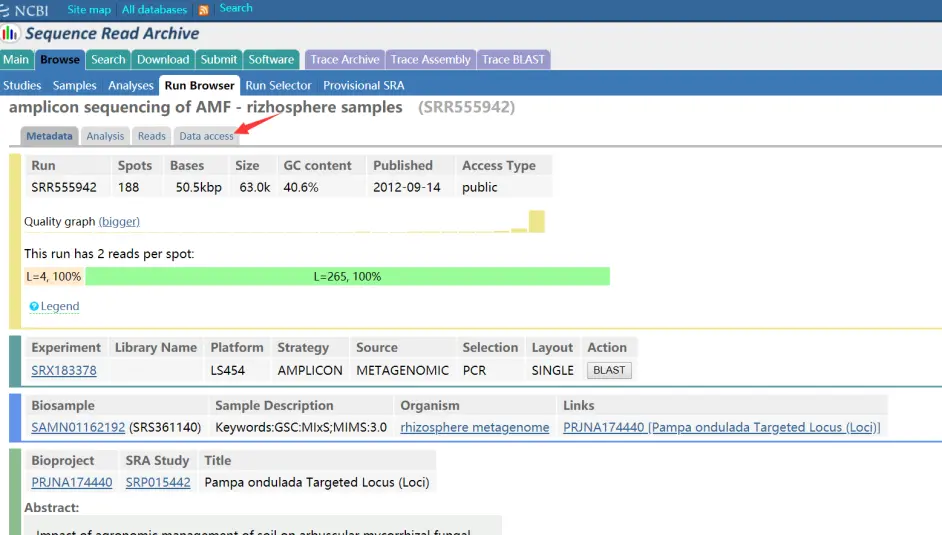
点开SRR555942之后的页面
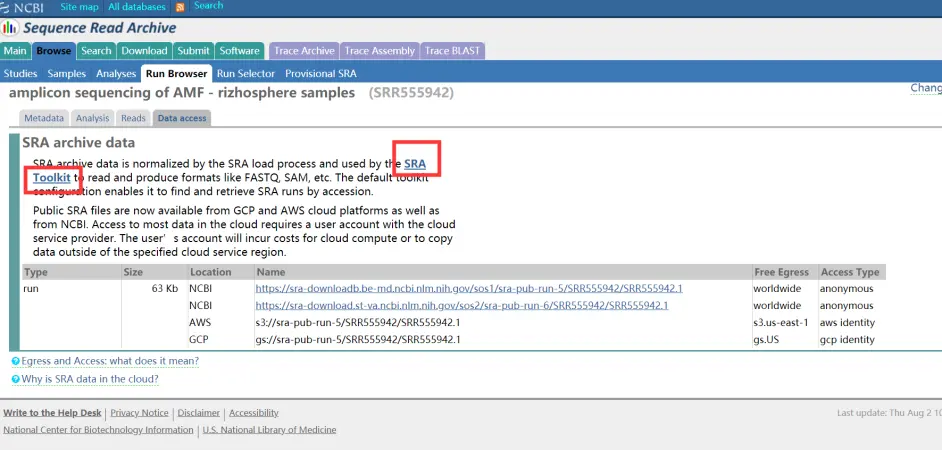
这里面介绍了它的基本信息,下载只需要点击Data access。
Run Browser : Browse : Sequence Read Archive : NCBI/NLM/NIH
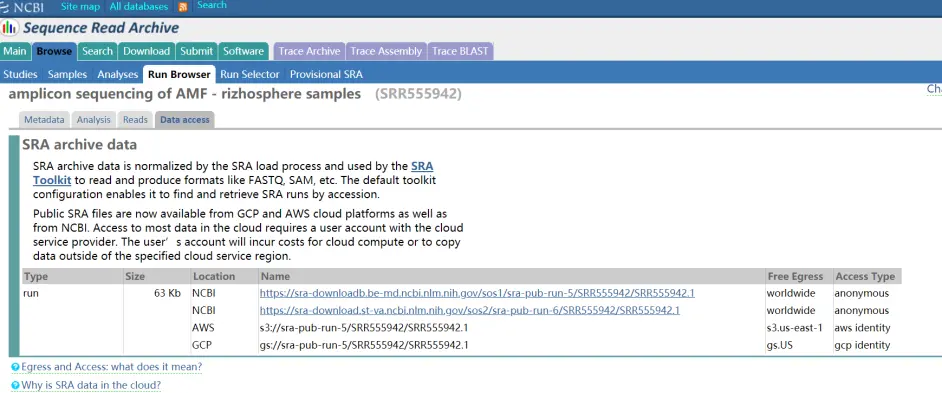
Data access页面
大小是63kb,这里面是包含序列信息和质量信息的。右边的链接随便点一个就可以下载了。下载下来之后是这么一个文件。
SRR文件
这个文件是不可以打开的,需要使用官方的fastq-dump将这个文件转换。
官方软件下载
点开之后
Toolkit Documentation : Software : Sequence Read Archive : NCBI/NLM/NIHhttps://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc
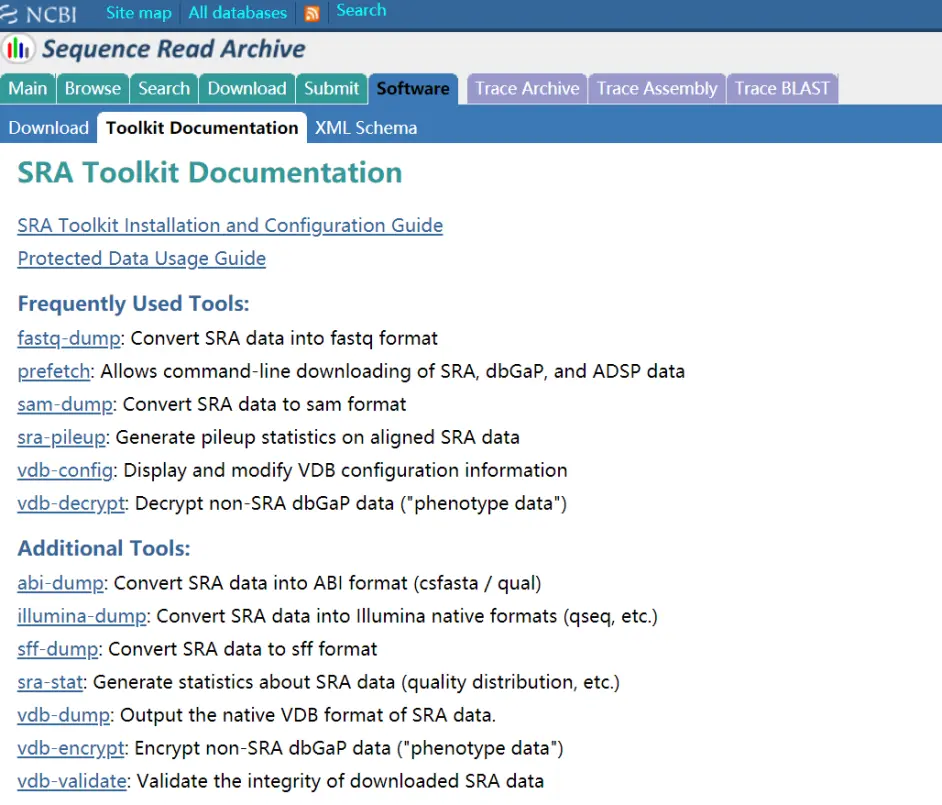
这里面提供了这么多软件,我们需要的是fastq-dump软件,prefetch这个软件可以直接从网上下载你要的序列,后面我会从视频中介绍。
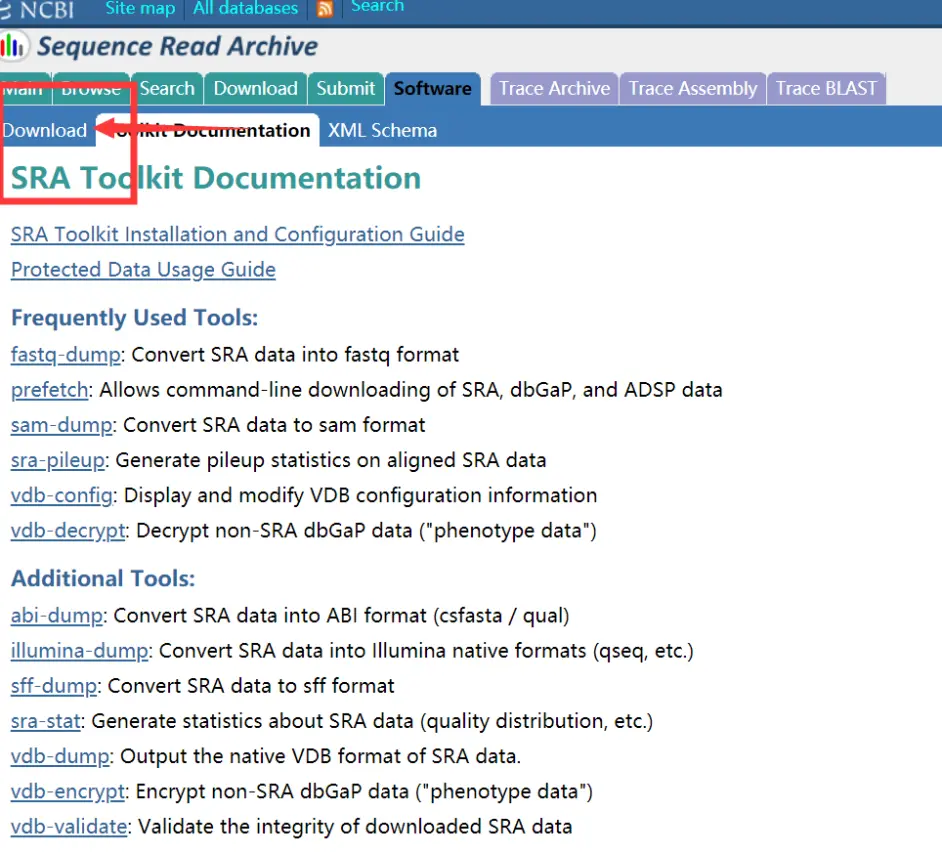
点击左边的Dowload
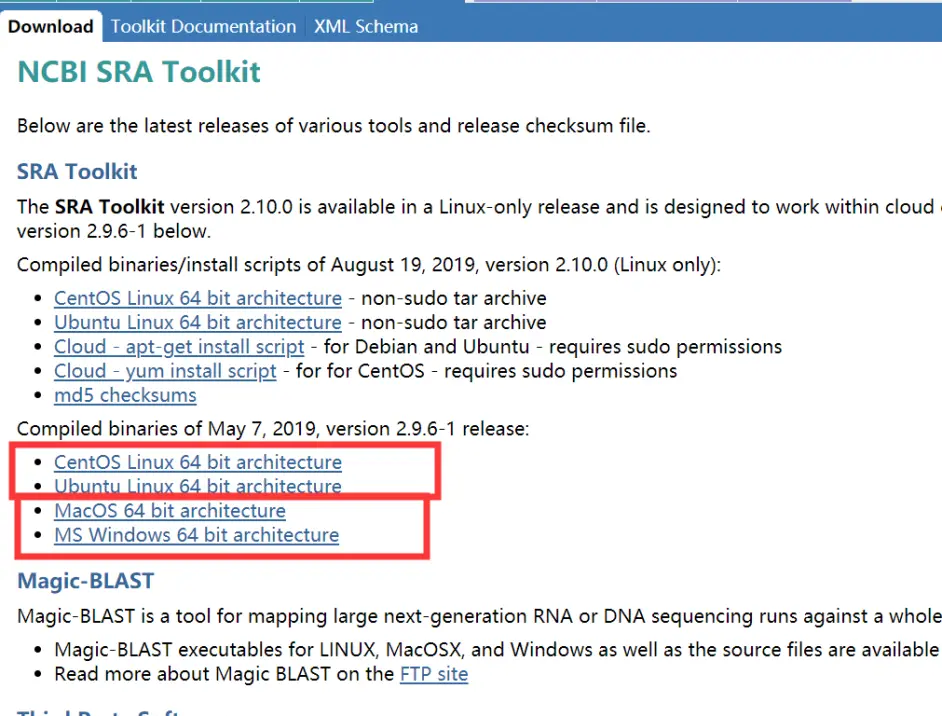
如果是Windows用户就下载最下面这个就可以了。
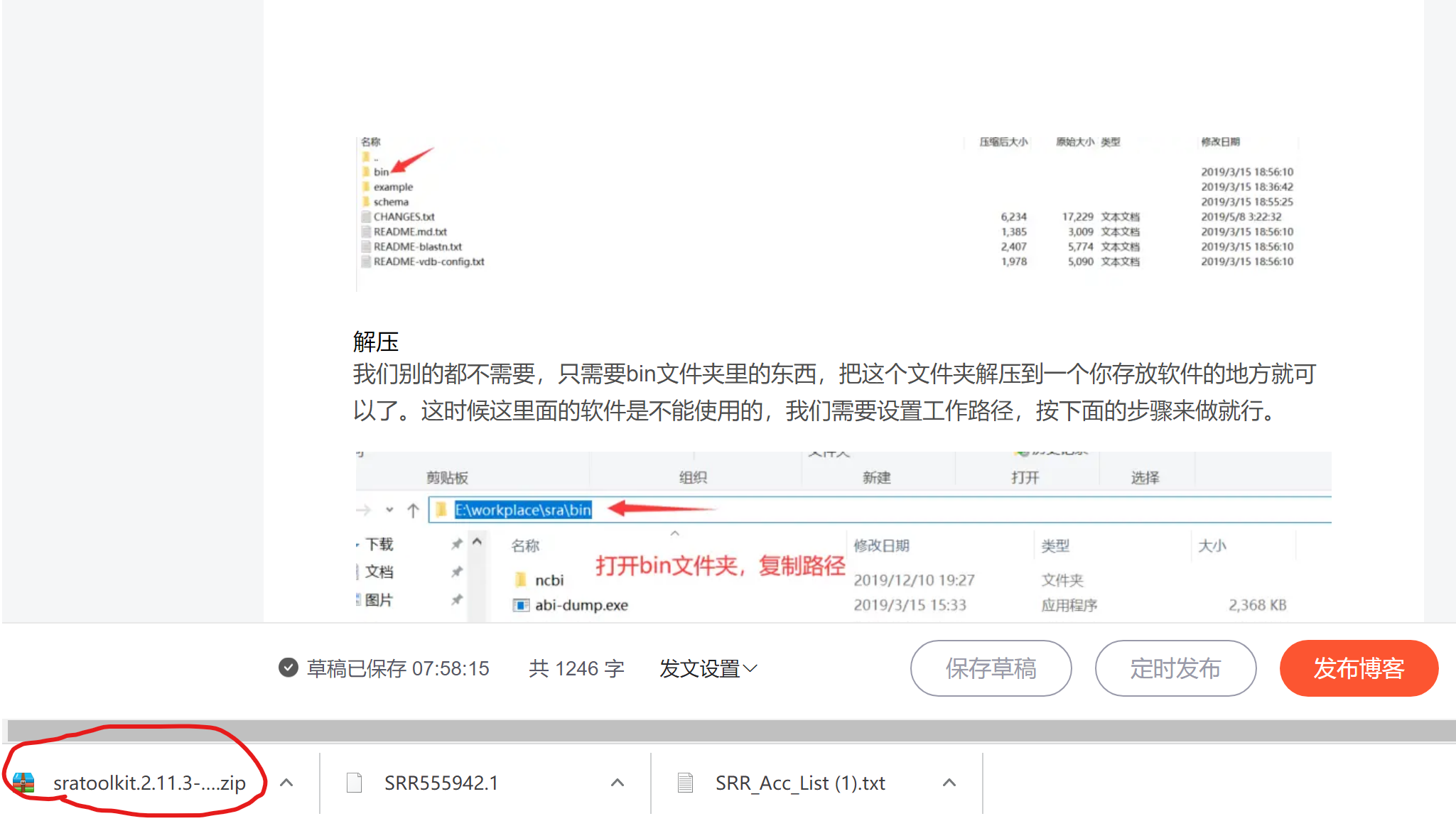

解压
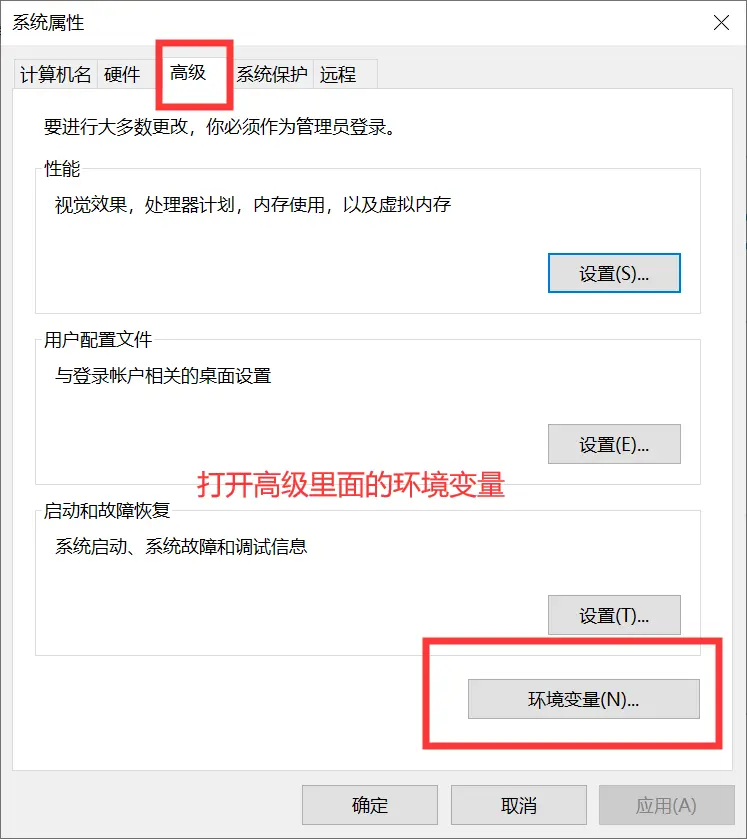
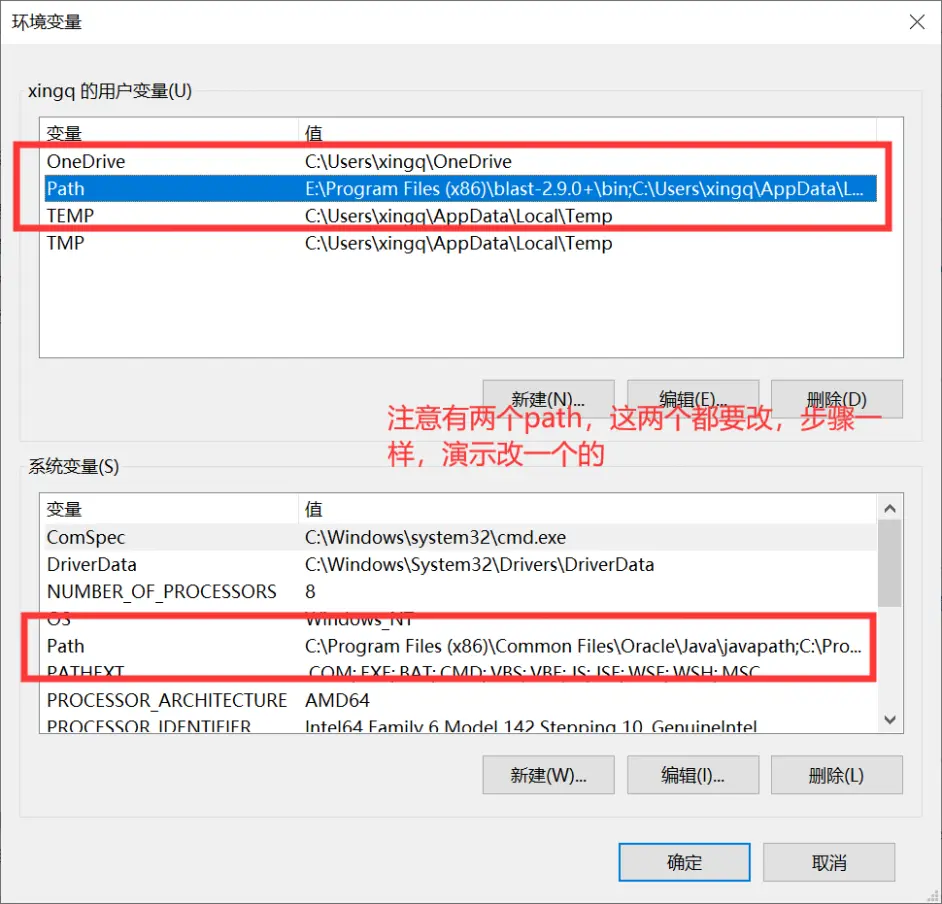
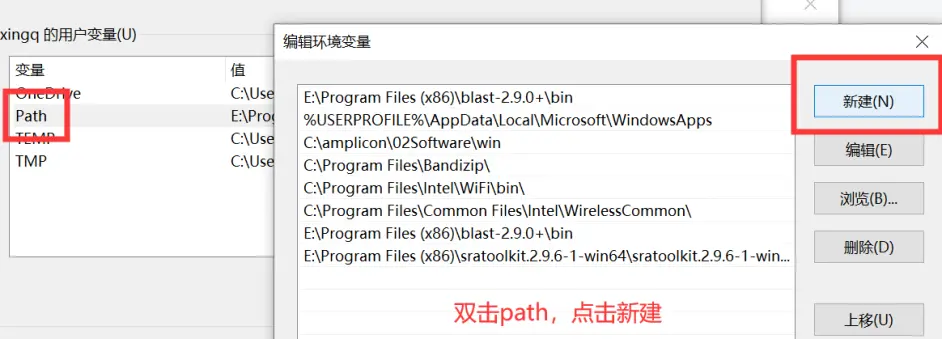
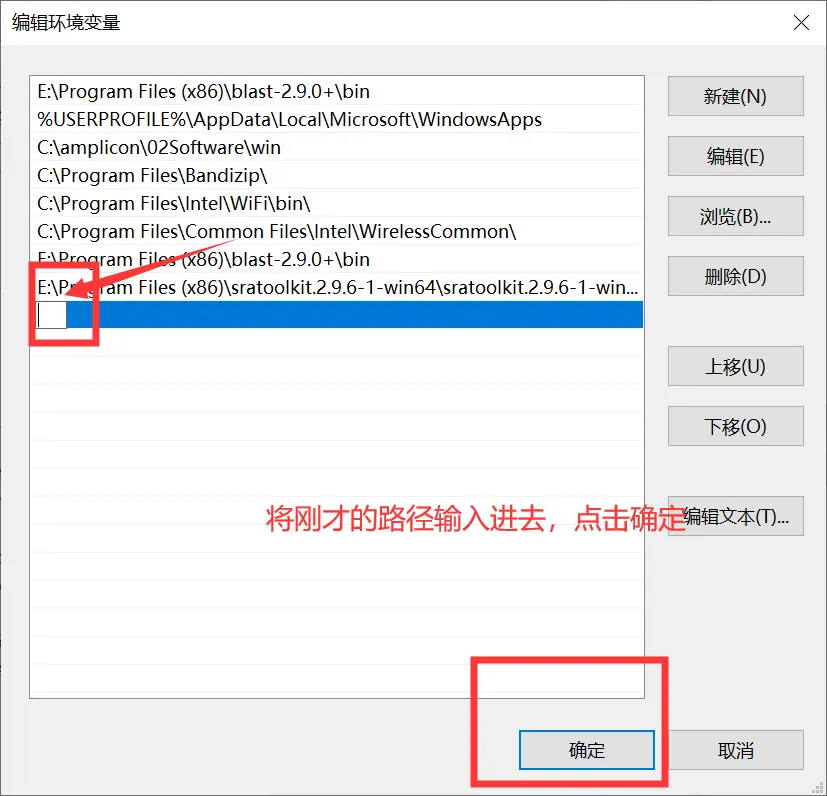
我们别的都不需要,只需要bin文件夹里的东西,把这个文件夹解压到一个你存放软件的地方就可以了。这时候这里面的软件是不能使用的,我们需要设置工作路径,按下面的步骤来做就行。
1
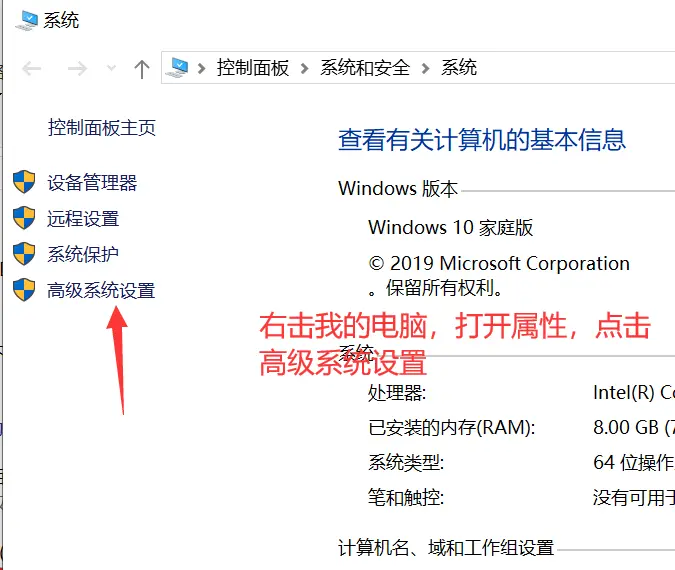
2
3
4
5
6
这时候应该就可以用了。在你刚刚保存SRR文件的路径上面输入CMD。
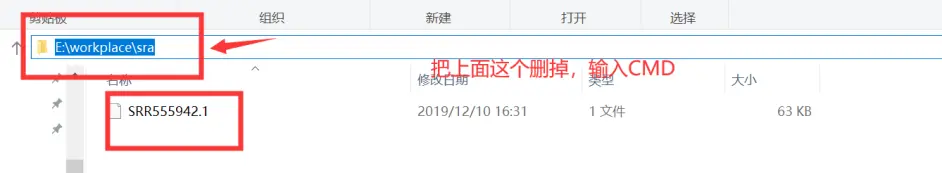
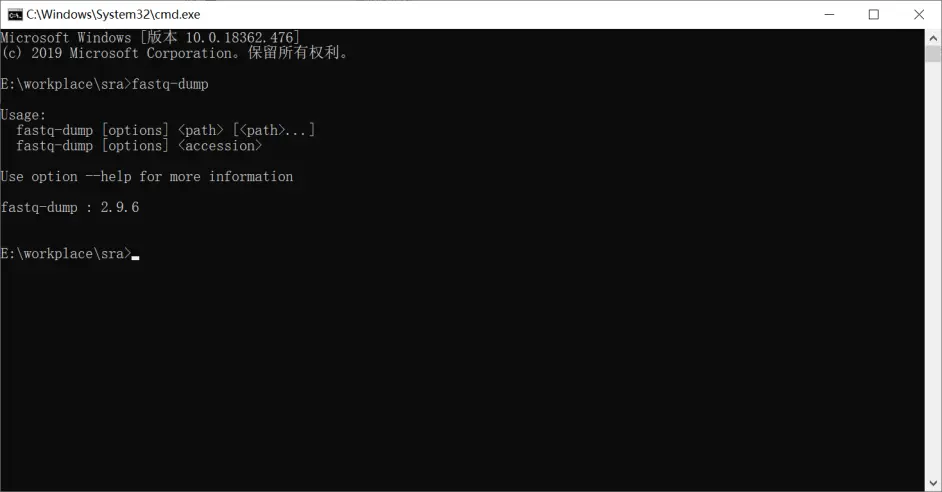
输入fastq-dump结果如下,说明路径设置成功了。
输入fastq-dump 你的ssr号
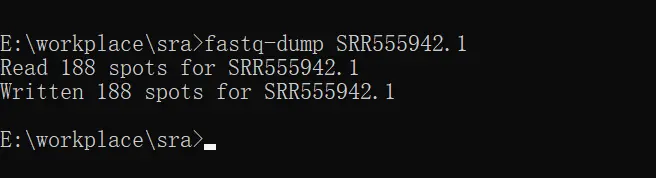
我的有188个序列。

生成的是fastq格式的文件,里面是包含序列质量的。

fastq文件
如果不想要序列质量,只想要序列,你可以输入 fastq-dump 你的文件 --fasta
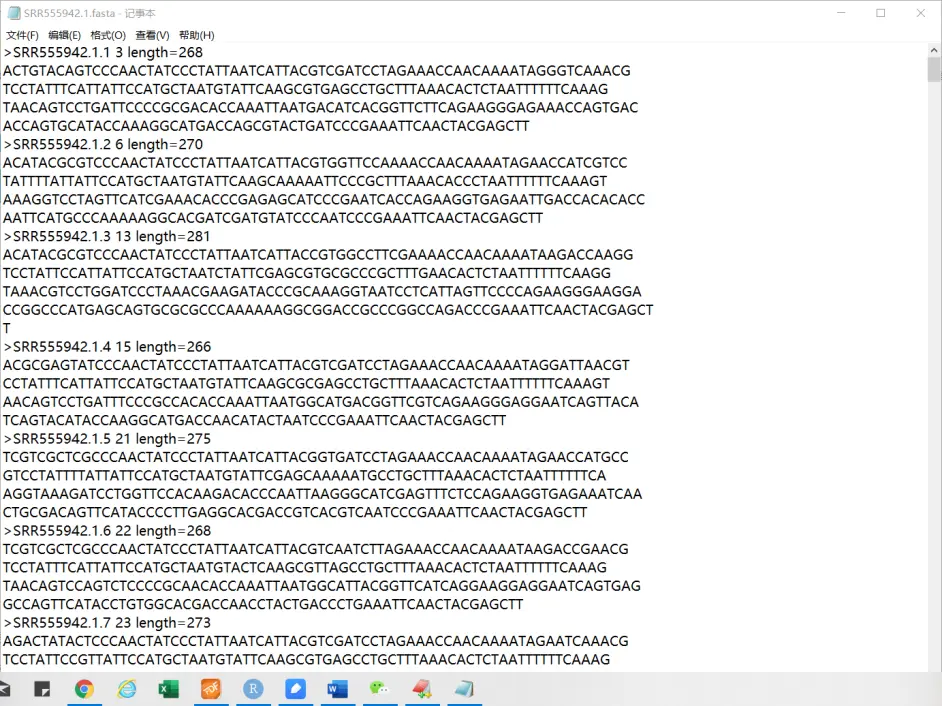
fasta文件
到此,序列下载结束。
后面我会通过视频来介绍如何用Rstudio来配置环境以及应用生物学常用的工具(不能算是软件吧,脚本吧)。
最后
以上就是闪闪咖啡豆最近收集整理的关于关于文献中二代测序数据下载(NCBI)的问题关于文献中二代测序数据下载(NCBI)的问题的全部内容,更多相关关于文献中二代测序数据下载(NCBI)内容请搜索靠谱客的其他文章。








发表评论 取消回复