写在前面
随着scRNAseq技术的普及,成本的降低,大家可以在公共数据库找到越来越多的datasets,但如何合并这些datasets就成为了一个大问题????,比较推荐的方法包括:????
Harmony;rliger;Seurat。
我们常见的2种应用场景就是:
3'和5'不同datasets的合并;- 整合只有部分重叠的
datasets,(举个栗子????:全血scRNAseq数据和3'PBMC数据的合并。
???? 在Seurat包中提供了一种叫canonical correlation analysis (CCA)的方法进行合并。
本期我们介绍一下如何通过Seurat包进行3'和5'不同datasets的整合。????
用到的包
rm(list = ls())
library(Seurat)
library(SeuratDisk)
library(SeuratWrappers)
library(patchwork)
library(harmony)
library(rliger)
library(RColorBrewer)
library(tidyverse)
library(reshape2)
library(ggsci)
library(ggstatsplot)
示例数据
这里我们提供1个3’ PBMC dataset和1个5’ PBMC dataset。????
matrix_3p <- Read10X_h5("./3p_pbmc10k_filt.h5",use.names = T)
matrix_5p <- Read10X_h5("./5p_pbmc10k_filt.h5",use.names = T)$`Gene Expression`
srat_3p <- CreateSeuratObject(matrix_3p,project = "pbmc10k_3p")
srat_5p <- CreateSeuratObject(matrix_5p,project = "pbmc10k_5p")
srat_3p
srat_5p
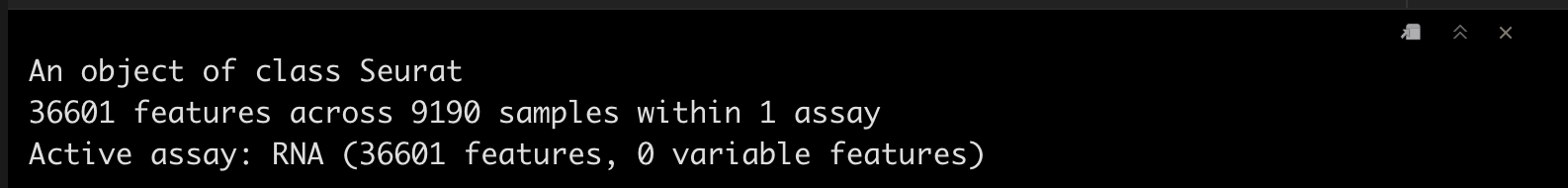
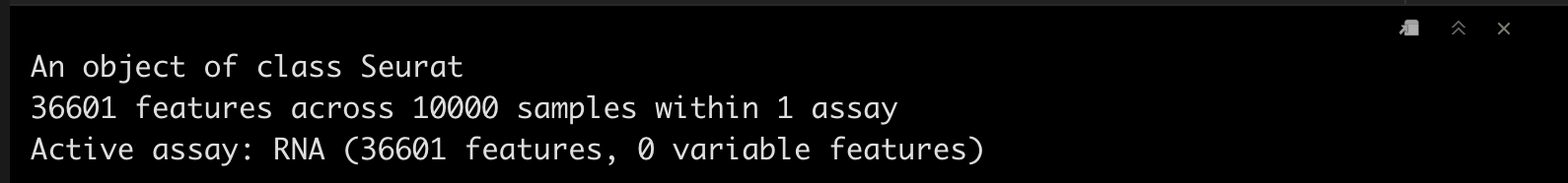
Note! 5' datset中还有一个assay,即VDJ data。????
线粒体与核糖体基因的计算
这里我们计算一下线粒体基因和核糖体基因比例,并进行可视化。????
srat_3p[["percent.mt"]] <- PercentageFeatureSet(srat_3p, pattern = "^MT-")
srat_3p[["percent.rbp"]] <- PercentageFeatureSet(srat_3p, pattern = "^RP[SL]")
srat_5p[["percent.mt"]] <- PercentageFeatureSet(srat_5p, pattern = "^MT-")
srat_5p[["percent.rbp"]] <- PercentageFeatureSet(srat_5p, pattern = "^RP[SL]")
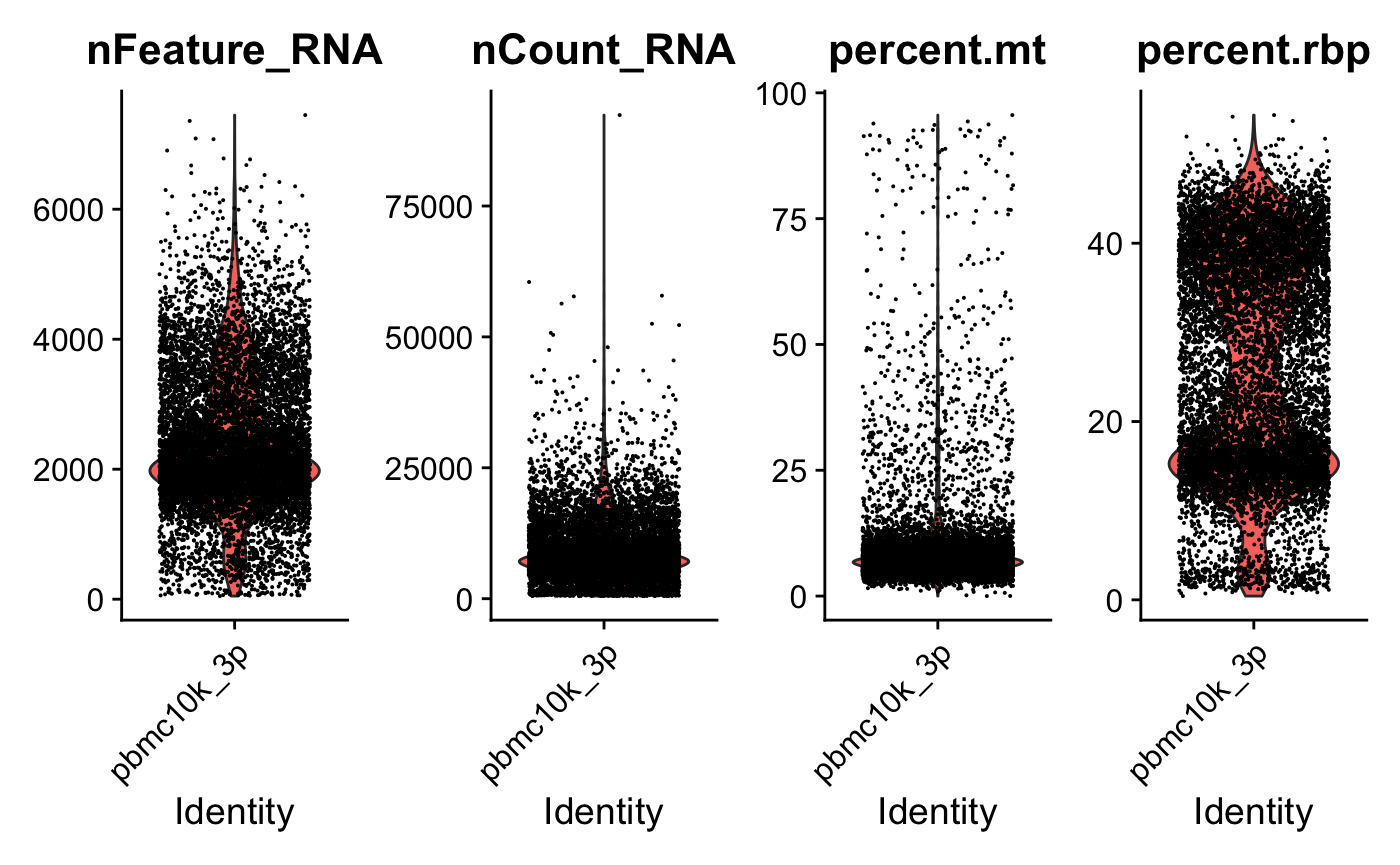
3’ dataset可视化 ????
VlnPlot(srat_3p, ncol = 4,
features = c("nFeature_RNA","nCount_RNA","percent.mt","percent.rbp"))
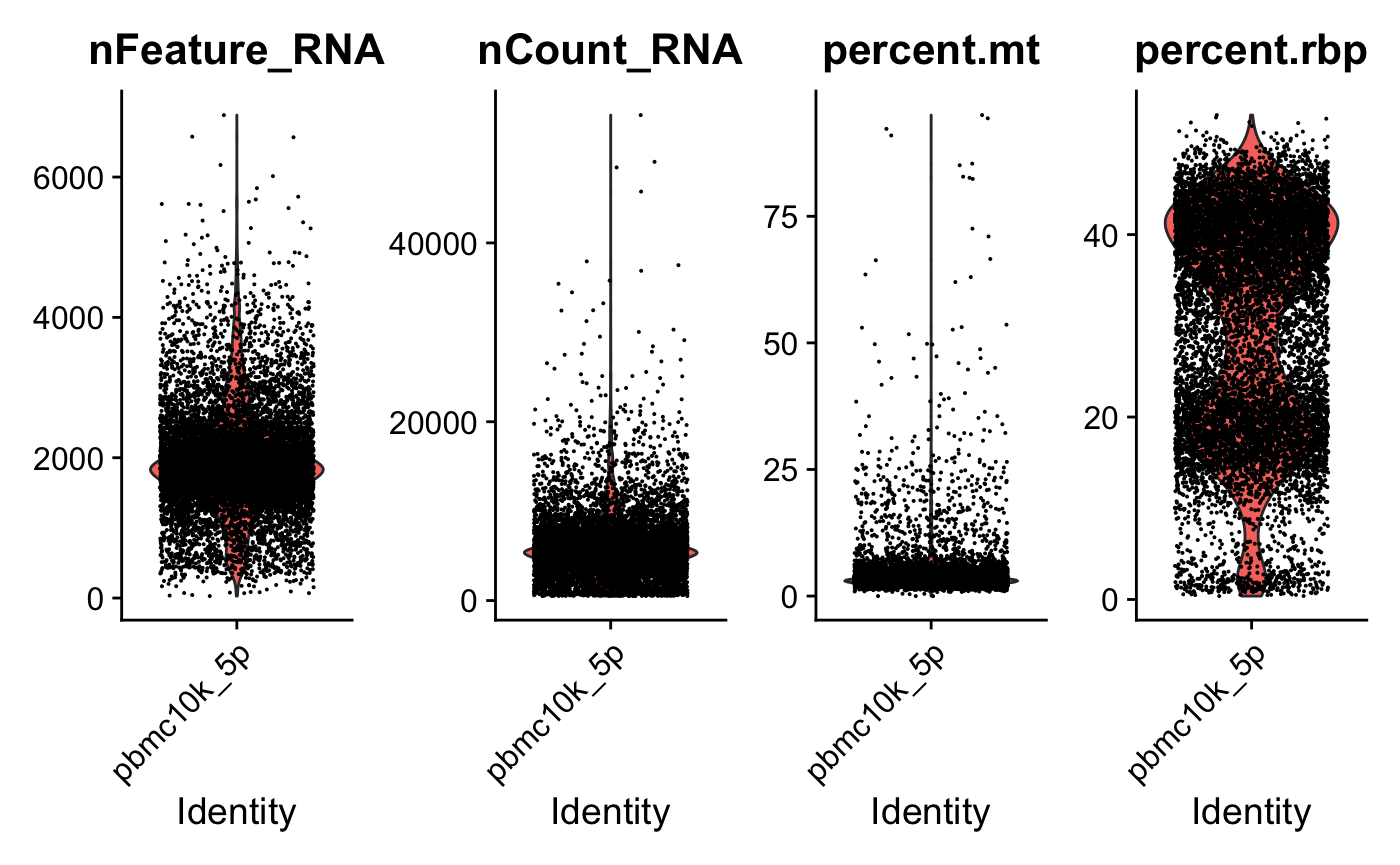
5’ dataset可视化 ????
VlnPlot(srat_5p, features = c("nFeature_RNA","nCount_RNA","percent.mt","percent.rbp"), ncol = 4)
接着我们看一下有多少基因名是一致的,我们使用的是Cell Ranger进行注释,背景文件为GRCh38-2020A。????
table(rownames(srat_3p) %in% rownames(srat_5p))

过滤
这里我们设置一下过滤条件,把死亡的细胞和doublets过滤掉。
srat_3p <- subset(srat_3p, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 15)
srat_5p <- subset(srat_5p, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 10)
合并
6.1 转为list
这里我们写个for循环,方便操作,同时完成Normalization和高变基因的选择。????
pbmc_list <- list()
pbmc_list[["pbmc10k_3p"]] <- srat_3p
pbmc_list[["pbmc10k_5p"]] <- srat_5p
for (i in 1:length(pbmc_list)) {
pbmc_list[[i]] <- NormalizeData(pbmc_list[[i]], verbose = F)
pbmc_list[[i]] <- FindVariableFeatures(pbmc_list[[i]], selection.method = "vst", nfeatures = 2000, verbose = F)
}
6.2 寻找合并Anchors
这一步耗时较长,16g的内存跑了大概5 min。????
pbmc_anchors <- FindIntegrationAnchors(object.list = pbmc_list, dims = 1:30)
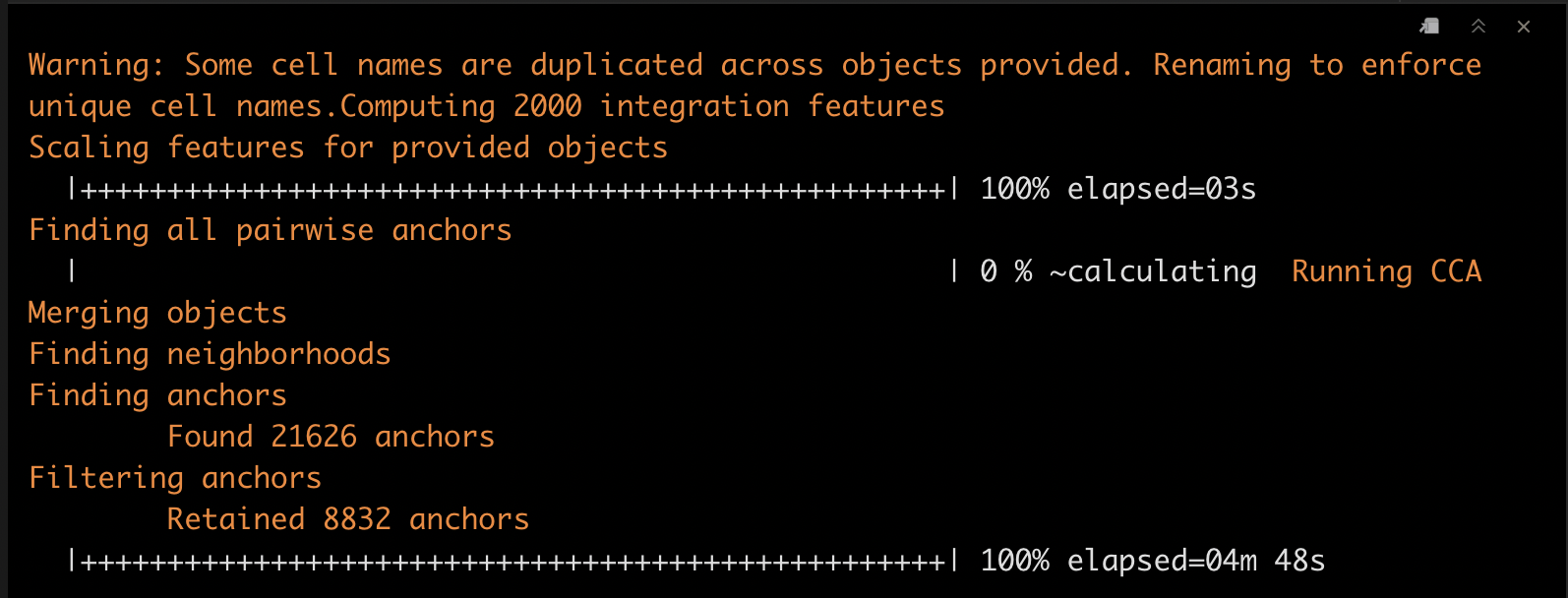
6.3 合并
pbmc_seurat <- IntegrateData(anchorset = pbmc_anchors, dims = 1:30)
rm(pbmc_list)
rm(pbmc_anchors)
合并前后的比较
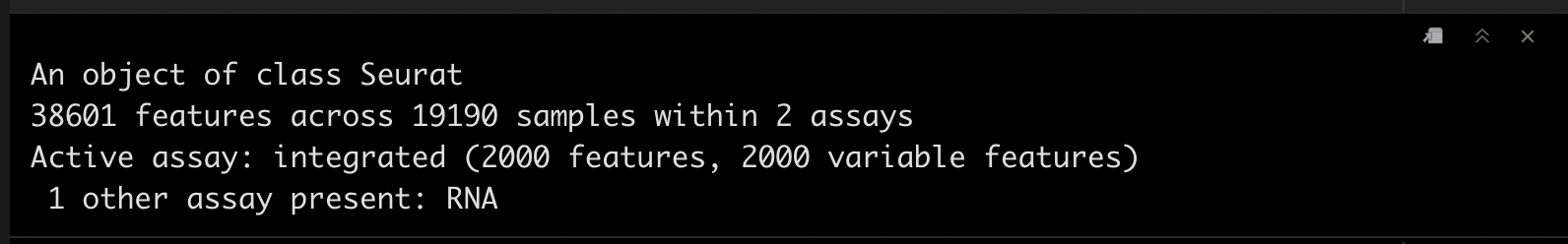
7.1 查看信息
这个时候我们看一下合并后的pbmc_seurat数据,我们拥有了两个assay,intergated和RNA。
pbmc_seurat
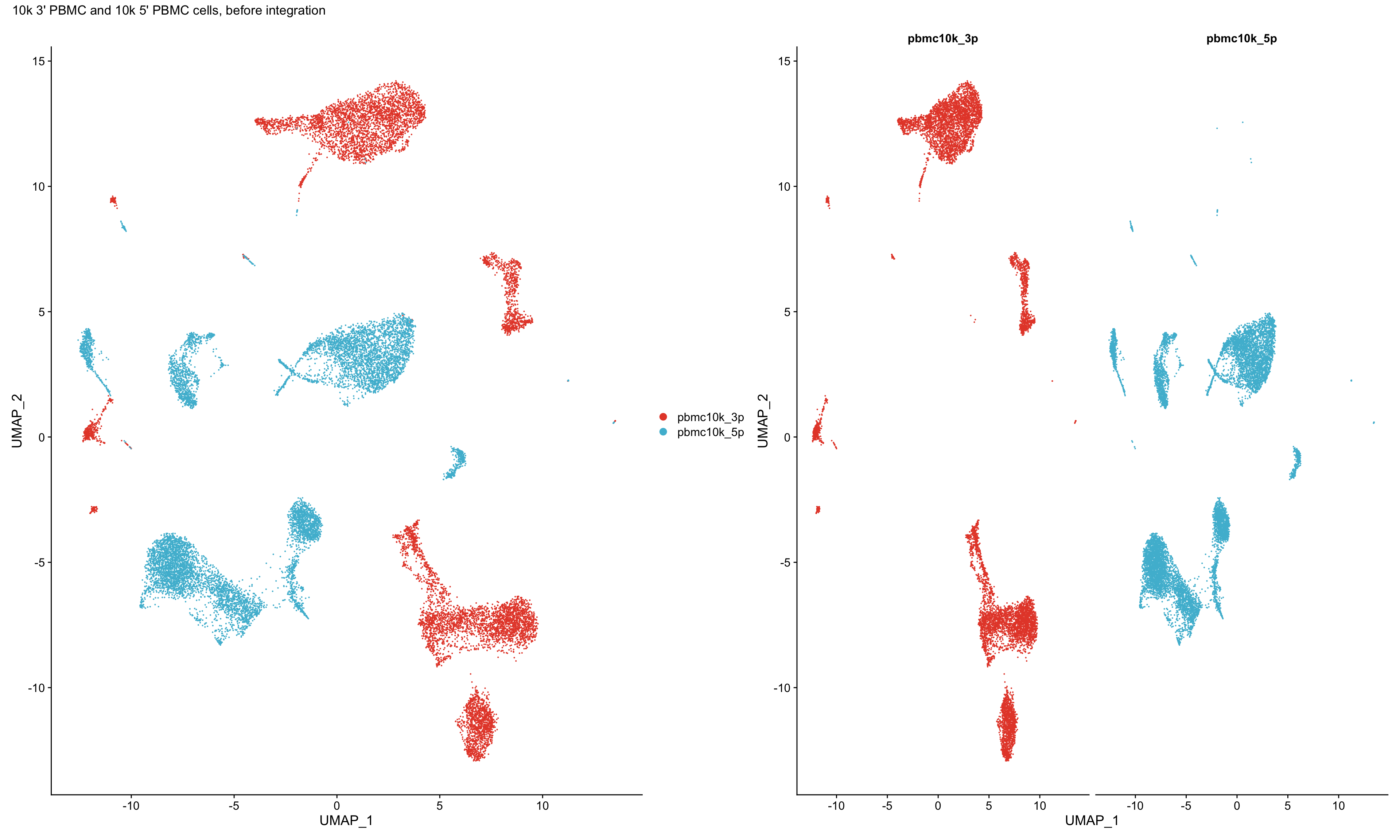
7.2 合并前
这里可以看到使用Seurat包的CCA方法合并前,PCA结果是明显分离的。
DefaultAssay(pbmc_seurat) <- "RNA"
pbmc_seurat <- NormalizeData(pbmc_seurat, verbose = F)
pbmc_seurat <- FindVariableFeatures(pbmc_seurat, selection.method = "vst", nfeatures = 2000, verbose = F)
pbmc_seurat <- ScaleData(pbmc_seurat, verbose = F)
pbmc_seurat <- RunPCA(pbmc_seurat, npcs = 30, verbose = F)
pbmc_seurat <- RunUMAP(pbmc_seurat, reduction = "pca", dims = 1:30, verbose = F)
### 可视化
p1 <- DimPlot(pbmc_seurat,reduction = "umap") +
scale_color_npg()+
plot_annotation(title = "10k 3' PBMC and 10k 5' PBMC cells, before integration")
p2 <- DimPlot(pbmc_seurat, reduction = "umap", split.by = "orig.ident") +
scale_color_npg()+
NoLegend()
p1 + p2
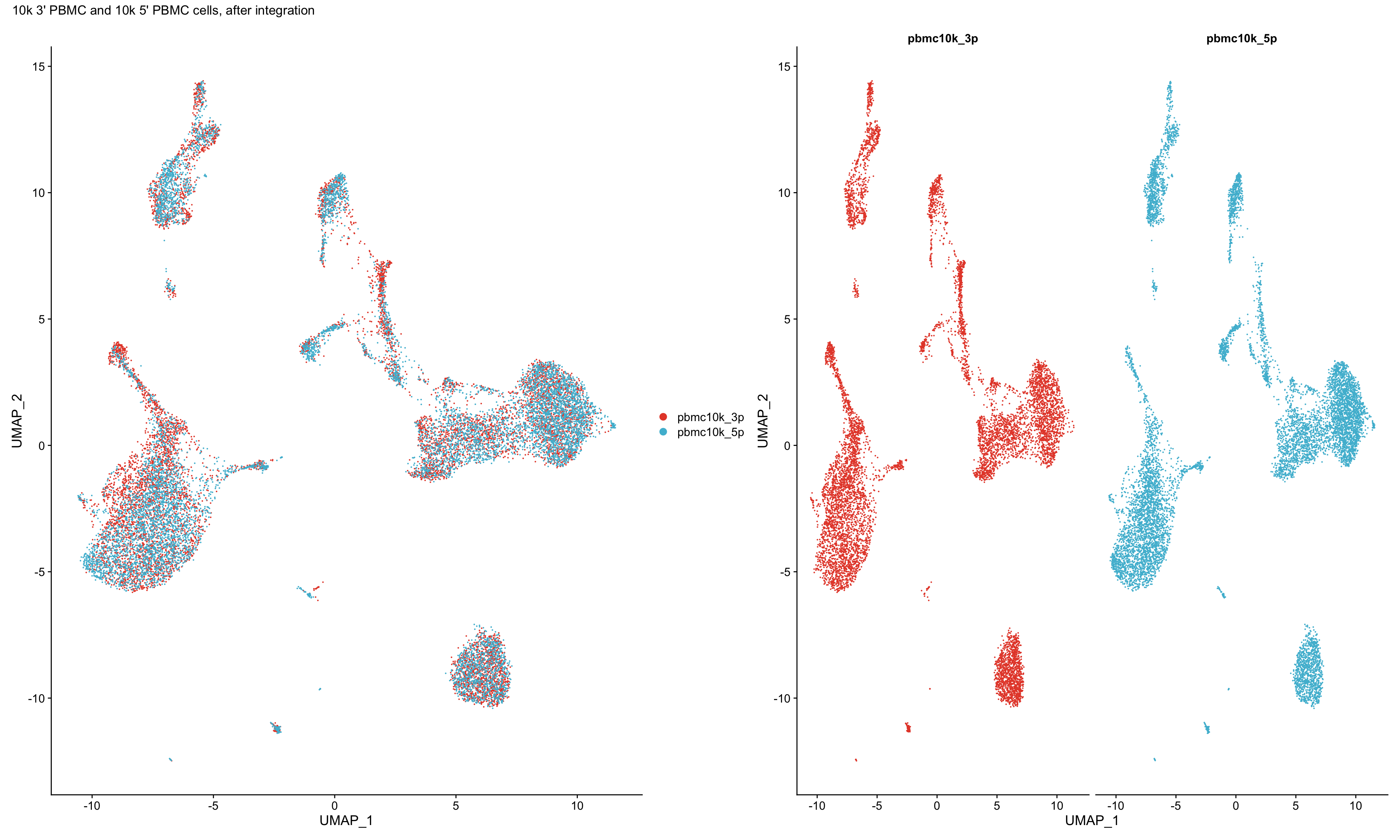
7.3 合并后
合并后,PCA结果完美重叠。
DefaultAssay(pbmc_seurat) <- "integrated"
pbmc_seurat <- ScaleData(pbmc_seurat, verbose = F)
pbmc_seurat <- RunPCA(pbmc_seurat, npcs = 30, verbose = F)
pbmc_seurat <- RunUMAP(pbmc_seurat, reduction = "pca", dims = 1:30, verbose = F)
### 可视化
p1 <- DimPlot(pbmc_seurat, reduction = "umap") +
scale_color_npg()+
plot_annotation(title = "10k 3' PBMC and 10k 5' PBMC cells, after integration")
p2 <- DimPlot(pbmc_seurat, reduction = "umap", split.by = "orig.ident") +
scale_color_npg()+
NoLegend()
p1 + p2
聚类
8.1 寻找clusters
合并完成后,我么就可以开始聚类,寻找clusters了。
pbmc_seurat <- FindNeighbors(pbmc_seurat, dims = 1:30, k.param = 10, verbose = F)
pbmc_seurat <- FindClusters(pbmc_seurat, verbose = F)
ncluster <- length(unique(pbmc_seurat[[]]$seurat_clusters))
mycol <- colorRampPalette(brewer.pal(8, "Set2"))(ncluster)
DimPlot(pbmc_seurat,
cols = mycol,
label = T,repel = T) +
NoLegend()
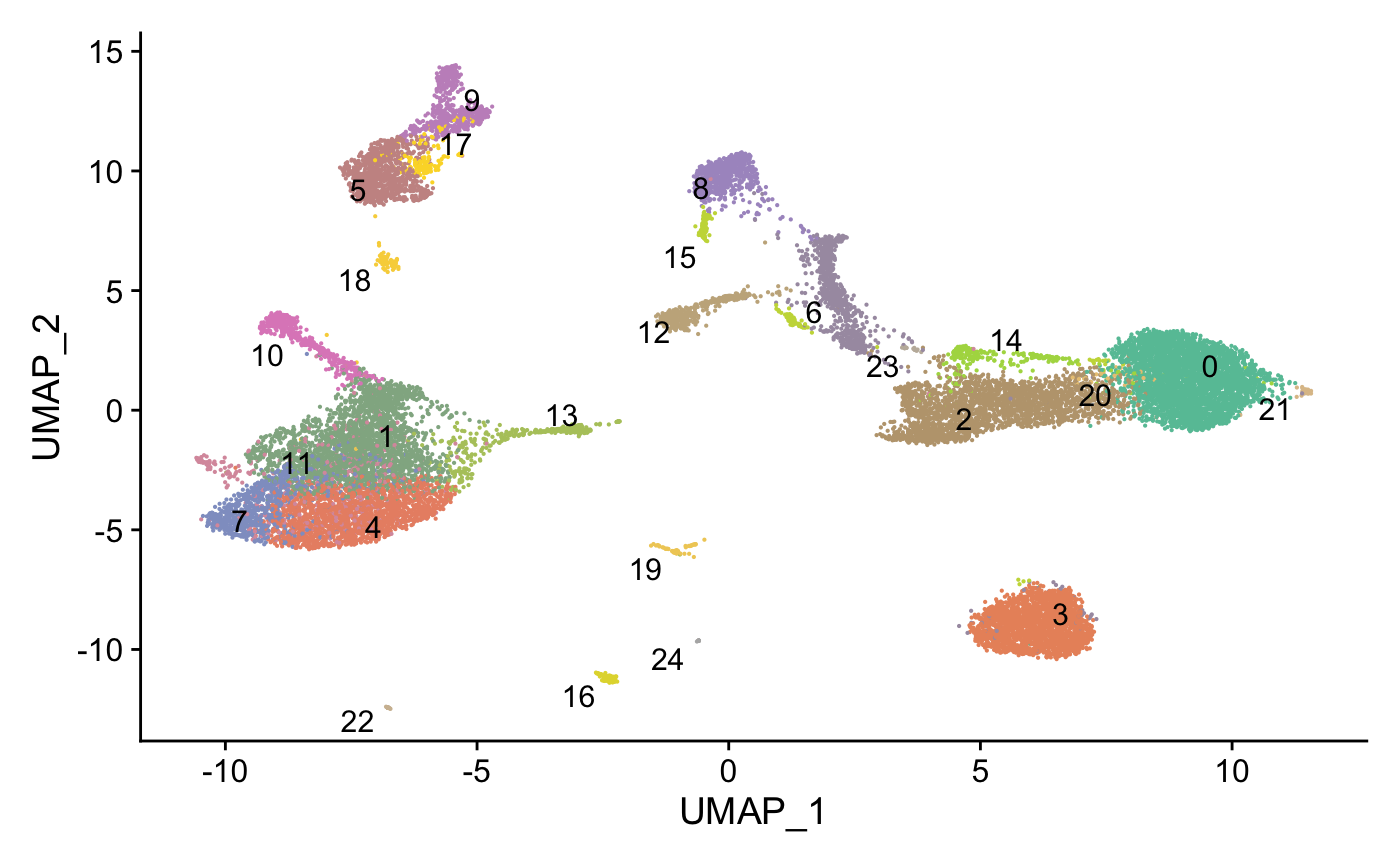
8.2 具体查看及可视化
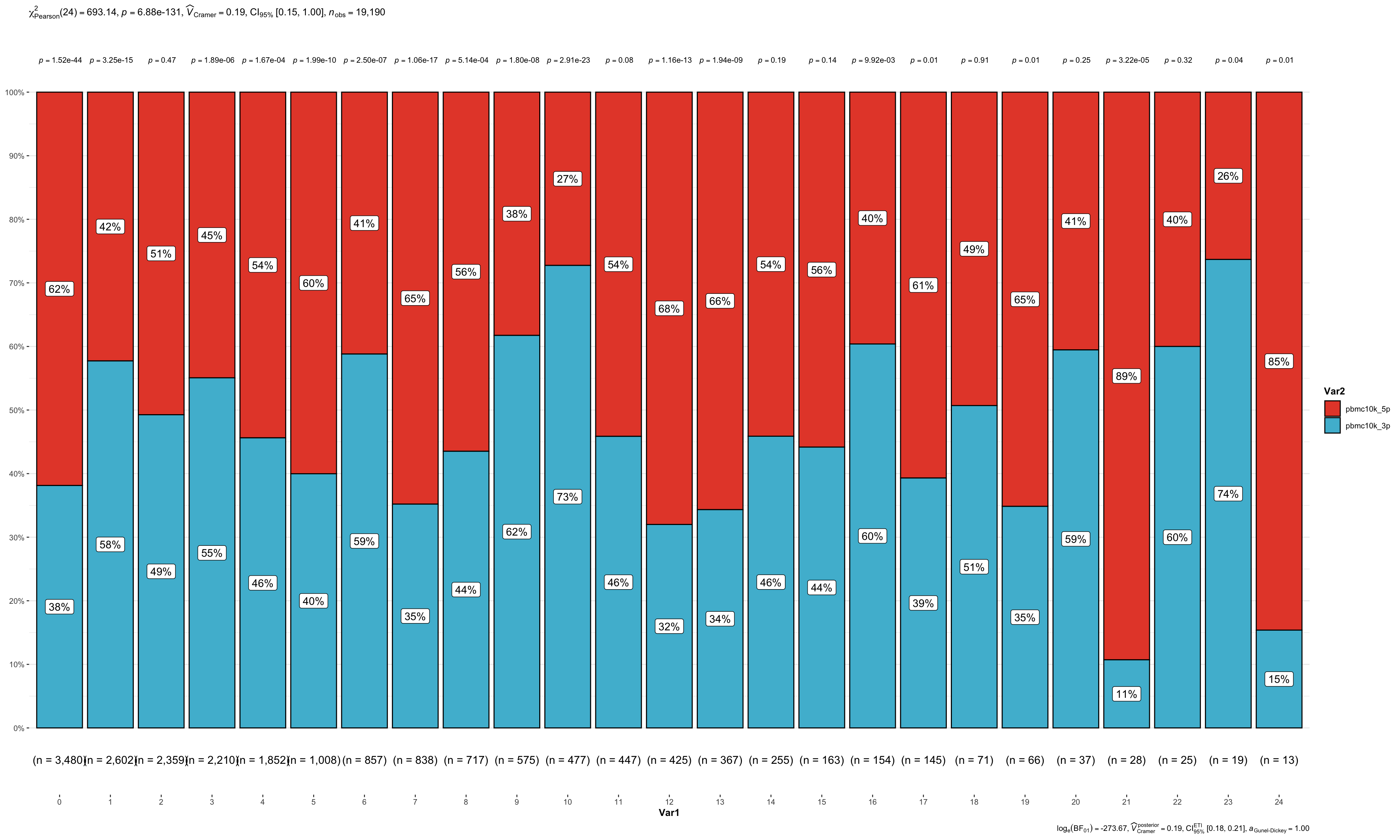
我们看下各个clusters在两个datasets各有多少细胞。
count_table <- table(pbmc_seurat@meta.data$seurat_clusters, pbmc_seurat@meta.data$orig.ident)
count_table
#### 可视化
count_table %>%
as.data.frame() %>%
ggbarstats(x = Var2,
y = Var1,
counts = Freq)+
scale_fill_npg()

需要示例数据的小伙伴,在公众号回复
Merge获取吧!点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
最后
以上就是彪壮草莓最近收集整理的关于Seurat | 超好用的单细胞测序数据合并(3‘和5‘数据合并)(一)的全部内容,更多相关Seurat内容请搜索靠谱客的其他文章。








发表评论 取消回复