Transcriptomic profiling of COS hiPSC-NPCs and hiPSC-neurons
Individuals with COS, as well as unaffected, unrelated healthy controls were recruited as part of a longitudinal study conducted at the National Institute of Health1 for available clinical information). This cohort is comprised of nearly equal numbers of cases and controls (Fig. 1a–c); 16 cases were selected representing a range of SZ-relevant CNVs, including 22q11.2 deletion, 16p11.2 duplication, 15q11.2 deletion, and NRXN1 deletion (2p16.3)1d; Supplementary Data 1).
Fig. 1
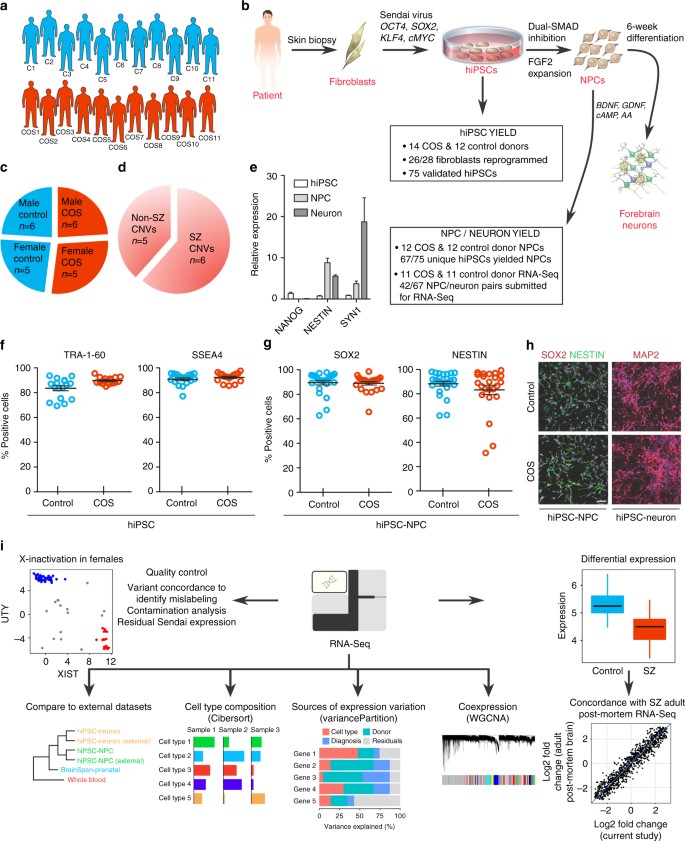
COS hiPSC cohort reprogramming and differentiation. a Validated hiPSCs (from 14 individuals with childhood-onset-schizophrenia (COS) and 12 unrelated healthy controls) and NPCs (12 COS; 12 control individuals) yielded 94 RNA-Seq samples (11 COS; 11 control individuals). b Schematic illustration of the reprogramming and differentiation process, noting the yield at each stage. c Sex breakdown of the COS-control cohort. d Breakdown of SZ-associated copy number variants in the 11 COS patients with RNA-Seq data. e Representative qPCR validation of NANOG, NESTIN, and SYN1 expression in hiPSCs (white bar), NPCs (light gray) and 6-week-old neurons (dark gray) from three individuals. f FACS analysis for pluripotency markers TRA-1-60 (left) and SSEA4 (right) in representative control (blue, n= 17) and COS (red, n= 16) hiPSCs. g FACS analysis for NPC markers SOX2 (left) and NESTIN (right) in control (blue, n= 34) and COS (red, n= 37) NPCs. h Representative images of NPCs (left) and 6-week-old forebrain neurons (right) from control (top) and COS (bottom). NPCs stained with SOX2 (red) and NESTIN (green); neurons stained with MAP2 (red). DAPI-stained nuclei (blue). Scale bar=50 μm. i Computational workflow showing quality control, integration with external data sets, computational deconvolution with Cibersort, decomposition multiple sources of expression variation with variancePartition, coexpression analysis with WGCNA, differential expression and concordance analysis
We used an integration free approach to generate genetically unmanipulated hiPSCs from COS patients (14 of 16 patients, 88% reprogrammed) and unrelated age- and sex-matched controls (12 of 12 controls, 100% reprogrammed) (Fig. 1b). Briefly, primary fibroblasts were reprogrammed by sendai viral delivery of KLF4, OCT4, SOX2, and cMYC; presumably clonal lines were picked and expanded 23–30 days following transduction. Following extensive immunohistochemistry, fluorescent activated cell sorting (FACS), quantitative polymerase chain reaction (qPCR) and karyotype assays to assess the quality of the hiPSCs (Fig. 1b, e, f), we selected two to three presumably clonal hiPSC lines per individual (n= 40 COS, n= 35 control, Table 1; Supplementary Data 1). A subset of these hiPSCs has been previously reported
Table 1 Number of individuals and cell lines at each step of experimental workflow
Using dual-SMAD inhibition, three to five forebrain hiPSC-NPC populations were differentiated from each validated hiPSC line via an embryoid body intermediate1g, h) (n = 32 COS, n= 35 control hiPSC-NPCs representing 67 unique hiPSC lines reprogrammed from 12 unique COS and 12 unique control individuals) were selected for further differentiation to 6-week-old forebrain neuron populations (Table 1
; Supplementary Data 2). We have previously demonstrated that hiPSC-NPCs can be directed to differentiate into mixed populations of excitatory neurons, inhibitory neurons and astrocytes
Because it required nearly 4 years to generate and differentiate all hiPSCs, hiPSC-NPCs, and hiPSC-neurons, it was not possible to fully apply standardized conditions across all cellular reprogramming and neural differentiations. Media reagents, substrates, and growth factors for fibroblast expansion, reprogramming, hiPSC differentiation, NPC expansion, and neuronal differentiation, as well as personnel and laboratory spaces, varied over time. Although individual fibroblast lines were reprogrammed and differentiated to hiPSC-NPCs in the order in which they were received, multiple randomization steps were introduced at the subsequent stages, particularly the thaw, expansion, and neuronal differentiation of validated hiPSC-NPCs in preparation for RNA sequencing (RNA-Seq) (see Supplementary Data 2 for available batch information). Only validated hiPSC-NPCs that yielded high quality populations of matched hiPSC-NPCs and hiPSC-neurons in one of three batches of thaws were used for RNA-Seq (Supplementary Data 1, 2).
RNA-Seq data were generated from 94 samples (n= 47 hiPSC-NPC, n= 47 hiPSC-neurons; n= 46 COS, n= 48 controls; representing 42 unique hiPSC lines reprogrammed from 11 unique COS and 11 unique control individuals) following ribosomal RNA (rRNA) depletion (Table 1; Supplementary Data 2). The median number of uniquely mapped read pairs per sample was 42.7 million, of which only a very small fraction were rRNA reads (Supplementary Fig. 1; Supplementary Data 3). In total 18,910 genes (based on ENSEMBL v70 annotations) were expressed at levels deemed sufficient for analysis (at least 1 CPM in at least 30% of samples); 11,681 were protein coding, 879 were lncRNA, and the remaining were of various biotypes (Supplementary Data 4).
Since six COS patients were selected based on CNV status, we examined gene expression in the regions affected by the CNVs. Despite the noise inherent to RNA-Seq and the high level of biologically driven expression variation in samples without CNVs, we identified corresponding hiPSC-NPC and hiPSC-neuron expression changes in some CNV regions (Supplementary Fig. 2).
In addition to SZ diagnosis-dependent effects, gene expression between hiPSC-NPCs and hiPSC-neurons was expected to vary as a result of technical
Addressing technical variation in RNA-Seq data
We implemented an extensive quality control pipeline to detect, minimize and account for many possible sources of technical variation (Fig. 1i). Samples were submitted and processed for RNA-Seq in only one batch; RNA isolation, library preparation, and sequencing were completed under standardized conditions at the New York Genome Center. Errors in sample mislabeling and cell culture contamination were identified, allowing us to correct sample labeling when possible and remove samples from further analysis when not. Batch effects in both tissue culture and RNA-Seq sample processing were corrected for and samples with aberrant X-inactivation
Expression patterns of genes on the sex chromosomes can identify the sex of each sample, confirm sample identity, and also measure the extent of X-inactivation in females. Using XIST on chrX and the expression of six genes on chrY (USP9Y, UTY, NLGN4Y, ZFY, RPS4Y1, TXLNG2P), this analysis identified 2 mislabeled males that show a female expression pattern and 15 female samples that have expression patterns intermediate between males and females (Supplementary Fig. 3A), consistent with either contamination or aberrant X-inactivation.
Samples with mislabeling and/or cross-individual contamination, whether during cell culture and/or RNA library preparation, were identified through genotype concordance analysis. VerifyBamIDn= 38 hiPSC-NPC, n= 38 hiPSC-neurons; n= 36 COS, n= 40 controls, from 10 unique COS and 9 unique control individuals) were validated for subsequent analysis (Table 1; Supplementary Data 2; Supplementary Fig. 3B).
Residual Sendai virus expression was assessed using Inchworm in the Trinity package2; Supplementary Fig. 4) showed evidence of persistent Sendai viral expression at > 1 count per million. Differential expression analysis identified 2768 genes correlated with Sendai expression at FDR
Overall, our rigorous bioinformatic strategy adjusted for technical variation and batch effects, eliminated spurious samples, and flagged samples that were contaminated or had aberrant X-inactivation. This extensive analysis was motivated by the high level of intra-donor expression variation (see below), and eliminating these factors as possible explanations for this expression variation ultimately improved our ability to resolve SZ-relevant biology in our data set.
COS RNA-Seq data cluster with existing data sets
To assess the similarity of our hiPSC-NPCs and hiPSC-neurons to other hiPSC studies (by ourselves and others), as well as to post-mortem brain, we compared our data set to publicly available hiPSC, hiPSC-derived NPCs/neurons, and post-mortem brain homogenate expression data sets (Fig. 2). Hierarchical clustering indicated that similarity in expression profiles is largely determined by cell type (Fig. 2a). hiPSC-NPC and hiPSC-neuron data sets were more similar to prenatal samples than postnatal or adult post-mortem samples2b) indicated that hiPSC-NPCs more resemble hiPSCs/hESCs than do hiPSC-neurons.
Fig. 2
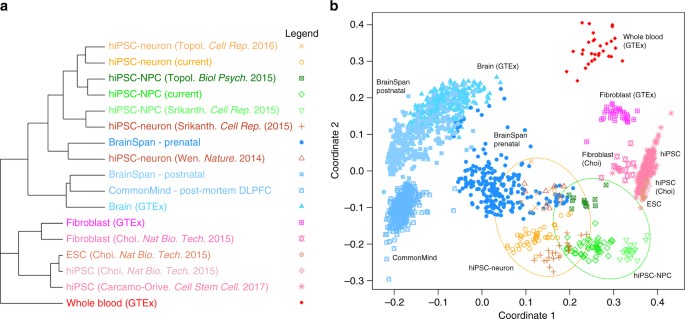
Cell type specificity of gene expression. a Summary of hierarchical clustering of 2082 RNA-Seq samples shows clustering by cell type. A pairwise distance matrix was computed for all samples, and the median distance between all samples in each category were used to create a summary distance matrix using to perform the final clustering. b Multidimensional scaling with samples colored as in a. hiPSC-NPCs from multiple studies are indicated in the green circle, and hiPSC-neurons from multiple studies are indicated in the orange circle
Genome-wide, hiPSC-NPCs and hiPSC-neurons express a common set of genes, so that expression differences between these cell types appear as changes in expression magnitude rather than activation of entirely different transcriptional modules (Supplementary Fig. 6). Yet this observation is also consistent with continuous variation in CTC, whereby the transcriptional signature of each cell type is present in each population at varying levels. Moreover, for both hiPSC-NPCs and hiPSC-neurons, genes that show high variance across donors in each cell type are enriched for brain eQTLs (Supplementary Fig. 7). Taken together, these two insights justified case-control comparisons within and between both hiPSC-NPCs and hiPSC-neurons.
Large heterogeneity in cell type composition
Given the substantial variability we observed between hiPSC-NPCs and hiPSC-neurons, even from the same individual (Supplementary Fig. 8), it seemed likely that inter-hiPSC and inter-NPC differences in differentiation propensity led to unique neural compositions in each sample. hiPSC-NPCs show extensive cell-to-cell variation in the expression of forebrain and neural stem cell markers
Bulk RNA-Seq analysis reflects multiple constituent cell types; therefore, we performed computational deconvolution analysis using CIBERSORT3). A reference panel of single-cell sequencing data from mouse brain
Fig. 3
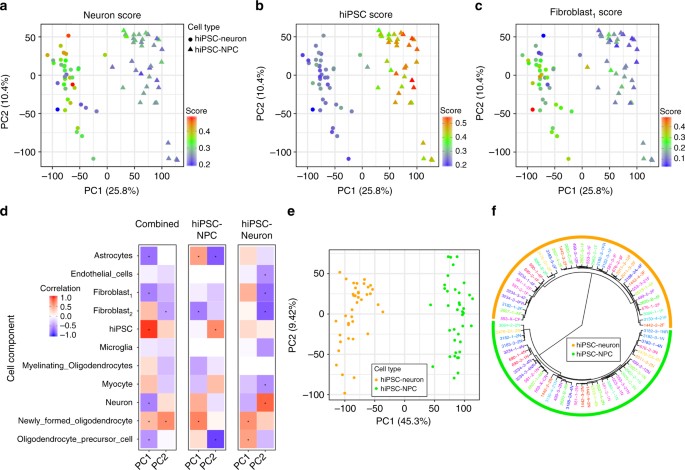
Variation in cell type composition contributes to gene expression variation. a–c Principal components analysis of gene expression data from hiPSC-NPCs (triangles) and hiPSC-neurons (circles) where samples are colored according to their cell type composition scores from cibersort for a neuron, b hiPSC, and c fibroblast1 components. Color gradient is shown on the bottom right of each panel. d Correlation between 11 cell type composition scores for the first two principal components of gene expression data from all samples, only hiPSC-NPCs, and only hiPSC-neurons. Red indicates a strong positive correlation with a principal component and blue indicates a strong negative correlation. Asterisks indicate correlations that are significantly different from zero with a p-value that passes the Bonferroni cutoff of 5% for 66 tests. e Principal components analysis of expression residuals after correcting for the two fibroblast cell type composition scores. f Hierarchical clustering of samples based on expression residuals after correcting for the two fibroblast cell type composition scores
Overlaying CTC scores on a principal component analysis (PCA) of the expression data indicates that hiPSC-NPCs and hiPSC-neurons separate along the first principal component (PC), explaining 25.8% of the variance, and that the cell types have distinct CTC scores (Fig. 3a–c). As expected, hiPSC-neuron samples had a higher neuron CTC score than hiPSC-NPCs (mean increase = 0.06, p
Not only is there significant overlap between fibroblast, mesenchymal and neural crest gene expression signatures (reviewed1 and fibroblast2 signatures only as a tool with which to assess the variability in differentiation quality; high values for the “fibroblast signature” may well imply the presence of non-fibroblast contaminant(s) such as neural crest and/or mesenchymal cells. Supplementary Fig. 10 plots the expression of key neural crest
The effect of CTC heterogeneity, likely due to the variation in differentiation efficiency, can be reduced by including multiple CTC scores in a regression model and computing the residuals. Using an unbiased strategy, we systematically evaluated which CTC score(s), when included in our model, most explained the variance in our samples. PCA on the residuals from a model including fibroblast1 and fibroblast2 CTC scores showed a markedly greater distinction between cell types, such that the first PC now explained 45.3% of the variance (Fig. 3e). Moreover, accounting for the CTC scores increased the similarity between the multiple biological replicates generated from the same donor and resulted in less intra-individual variation within each cell type (Fig. 3f, Supplementary Fig. 11). Finally, accounting for CTC was necessary in order to see concordance with one of the adult post-mortem cohorts (see below).
Characterizing known sources of expression variation
As discussed above, gene expression (in our data set and others) is impacted by a number of biological and technical factors. By properly attributing multiple sources of expression variation, it is possible to (partially) correct for some variables. To decompose gene expression into the percentage attributable to multiple biological and technical sources of variation, we applied variancePartition4). For each gene we calculated the percentage of expression variation attributable to cell type, donor, diagnosis, sex, as well as CTC scores for both fibroblast sets. All remaining expression variation not attributable to these factors was termed residual variation. The influence of each factor varies widely across genes; while expression variation in some genes is attributable to cell type, other genes are affected by multiple factors (Fig. 4a). Overall, and consistent with the separation of hiPSC-NPCs and hiPSC-neurons by the first PC, cell type has the largest genome-wide effect and explained a median of 13.3% of the observed expression variation (Fig. 4b). Expression variation due to diagnosis (i.e., between SZ and controls) had a detectable effect in a small number of genes. Meanwhile, variation across the sexes was small genome-wide, but it explained a large percentage of expression variation for genes on chrX and chrY. Technical variables such as hiPSC technician, hiPSC date, NPC generation batch, NPC technician, sample name, NPC thaw and RIN explained little expression variation (Supplementary Fig. 12), especially compared to technical effects observed in previous studies
Fig. 4
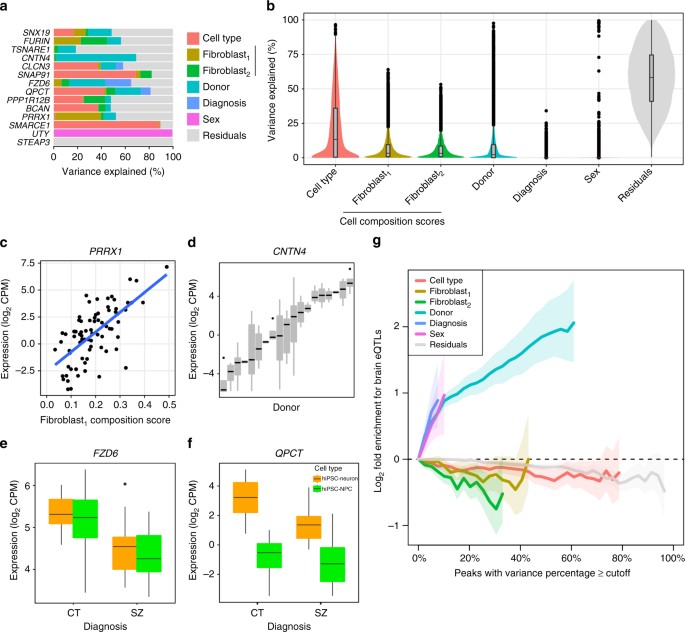
Decomposing expression variation into multiple sources. a Expression variance is partitioned into fractions attributable to each experimental variable. Genes shown include genes of known biological relevance to schizophrenia and genes for which one of the variables explains a large fraction of total variance. b Violin plots of the percentage of variance explained by each variable over all the genes. c–f Expression of representative genes stratified by a variable that explains a substantial fraction of the expression variation. c
PRRX1 plotted as a function of the fibroblast1 cell type composition score. d
CNTN4 stratified by donor. e
FZD6 stratified by disease status and cell type. f
QPCT stratified by disease status and cell type. g Genes that vary most across donors are enriched for brain cis-eQTLs. Fold enrichment (log2) for the 2000 top cis-eQTLs discovered in post mortem dorsolateral prefrontal cortex data generated by the CommonMind Consortiumx-axis. Shaded regions indicate the 90% confidence interval based on 10,000 permutations of the variance fractions. Enrichments are shown on the x-axis until less that 100 genes pass the cutoff
Variation attributable to cell type heterogeneity across the CTC scores had a larger median effect than the variation across the 22 donors (fibroblast1: 3.3%, fibroblast2: 3.2%). The median observed variation across donor is 2.2%, substantially lower than reported in other data sets from hiPSCsp
The percentage of expression variation explained by each factor has a specific biological interpretation. PRRX1 is known to function in fibroblasts1 CTC score explains 38.3% of expression variant in this gene (Fig. 4c). Expression of CNTN4 is driven by an eQTL in brain tissue that corresponds a risk locus for SZCNTN4 has 67.4% expression variation across donors suggesting that this variation is driven by genetics (Fig. 4d). Genes that vary across diagnosis correspond to differentially expressed genes, including FZD6, a WNT signaling gene linked to depression4e) and QPCT, a pituitary glutaminyl-peptide cyclotransferase that has been previously associated with SZ4f).
Genes that vary across donors were enriched for eQTLs detected in post-mortem brain tissue4g), meaning that observed inter-individual expression variation reflected genetic regulation of expression. Conversely, genes with expression variation attributable to cell type (CTC scores) are either neutral or depleted for genes under genetic control, indicating that variation in CTC was either stochastic or epigenetic, but did not reflect genetic differences between individuals. Finally, the high percentage of residual variation not explained by factors considered here suggests that there are other uncharacterized sources of expression variation, including stochastic canalization effects or unexplained variation in CTC.
WGCNA analysis identifies modules enriched for SZ and CTC
Genes with similar functions are known to share regulatory mechanisms and so are often coexpressed5, Supplementary Data 7). Genes were clustered into modules of a minimum of 20 genes, and each module was labeled with a color (Supplementary Fig. 14). Genes that did not form strong clusters were assigned to the gray module. Analysis was performed separately in hiPSC-NPCs and hiPSC-neurons; each module was evaluated for enrichment of genes for multiple biological processes. Many modules were highly enriched for genes that were significantly correlated with CTC scores at FDR
Fig. 5
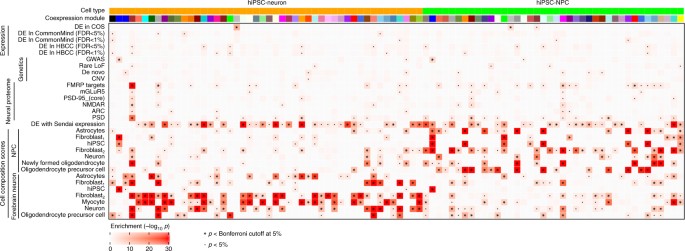
Clustering of genes into coexpression modules reveals module-specific enrichments. Enrichment significance (−log10
p-values from hypergeometric test) are shown for coexpression modules from hiPSC-NPCs and hiPSC-neurons. Each module is assigned a color and only modules with an enrichment passing the Bonferroni cutoff in at least one category is shown. Enrichments are shown for gene sets from RNA-Seq studies of differential expression between schizophrenia and controls; genetic studies of schizophrenia, neuronal proteomep-values passing the 5% Bonferroni cutoff are indicated by ‘*’, and p-values
Differential expression between COS and control hiPSC-NPCs and hiPSC-neurons
The central objective of this study was to determine if a gene expression signature of SZ could be detected in an experimentally tractable cell culture model (Fig. 6). Due to the “repeated measures” study design where individuals are represented by multiple independent hiPSC-NPC and hiPSC-neuron lines, we used a linear mixed model by applying the duplicateCorrelation function in our limma/voom analysis
Fig. 6
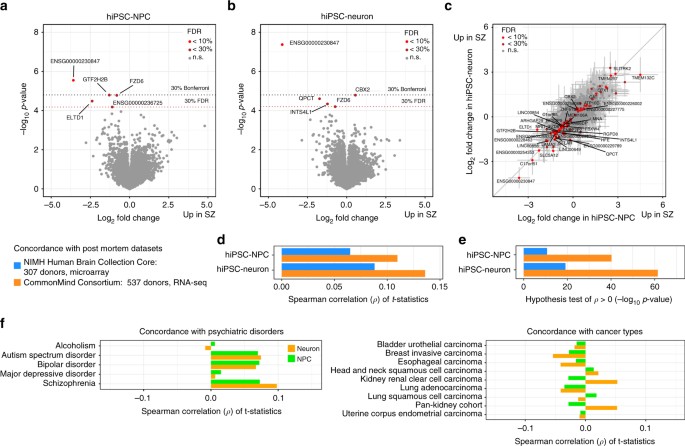
Differential expression between schizophrenia and controls. a, b Volcano plot showing log2 fold change between cases and controls and the –log10
p-value for each gene in a hiPSC-NPC and b hiPSC-neuron samples. Genes are colored based on false discovery rate: light red (FDR
p-values from a one-sided hypothesis test for the Spearman correlation coefficients from d being greater than zero. f Concordance of t-statistics with differential expression results from case-control analysis of five psychiatric diseases
Differential expression analysis between cases and controls in hiPSC-NPCs (Fig. 6a) identified 1 gene with FDR
While plausible candidates such as FZD6 and QPCT were differentially expressed, gene set enrichment testing did not implicate a coherent set of pathways (Supplementary Data 9). As SZ is a highly polygenic disease and this data set is underpowered due to the small sample size2 fold changes (Fig. 6c). Moreover, no genes had log2 fold changes that were statistically different in the two cell types, although we were underpowered to detect such differences.
Overall, our differential expression analysis demonstrated that case-control hiPSC-based cohorts remain under-powered to resolve biologically coherent SZ-associated processes. Nonetheless, the concordance in the disease signature identified in hiPSC-NPCs and hiPSC-neurons implies that future studies could focus on just one cell type.
Concordant differential gene expression with post-mortem data sets
While it is well-understood that all hiPSC-based studies of SZ remain under-powered due to small sample sizes and polygenic disease architecture, what is less appreciated is that post-mortem approaches are similarly constrained. Using allele frequencies from the Psychiatric Genetics Consortium data, the median number of subjects needed to obtain 80% power to resolve genome-wide expression differences in SZ cases was estimated to be ~28,500, well beyond any existing data set
The Spearman correlation between our hiPSC-NPC results and the CMC and NIMH HBCC results were 0.108 and 0.0661, respectively; for the hiPSC-neurons results, the correlations were 0.134 and 0.0896, respectively (Fig. 6d, Supplementary Figs. 15 and 16). These correlations were highly statistically significant (Fig. 6e) for both hiPSC-NPCs: p
While the concordance with CMC was observed when correcting for any set of CTC scores (or none), the concordance with HBCC was only apparent when correcting for the fibroblast1 CTC score (Supplementary Fig. 17). This illustrates the importance of accounting for CTC and the fact that concordance can be obscured by biological sources of expression variation. The genes for which the differential expression signal was boosted by accounting for the fibroblast1 score were enriched for brain and synaptic genesets, including specific biological functions such as FMRP and mGluR5 targets (Supplementary Figs. 18 and 19).
Given the degree of concordance in the SZ differentially expressed genes between the hiPSC-NPCs, hiPSC-neurons, CMC and NIMH HBCC data sets (Fig. 6d, e), the lack of enrichment of the CMC or NIMH HBCC differentially expressed genes in the “gray module” of our coexpression analysis (Fig. 5) is noteworthy. Although the concordance and coherence of the signal between hiPSC-NPCs and hiPSC-neurons with two post-mortem data sets was relatively low, we believe this reflects the small sample size and low power of our current study and predict that both will increase with expanding sample sizes in future studies.
最后
以上就是包容海燕最近收集整理的关于signature=a702a44d1f46acac87955d98d4ef1534,Transcriptional signatures of schizophrenia in hiPSC-deri...的全部内容,更多相关signature=a702a44d1f46acac87955d98d4ef1534,Transcriptional内容请搜索靠谱客的其他文章。








发表评论 取消回复